


A Ligand-mediated Hydrogen Bond Network Required for the Activation of the Mineralocorticoid Receptor*,[boxs]
- Randy K. Bledsoe‡§,
- Kevin P. Madauss¶,
- Jason A. Holt∥,
- Christopher J. Apolito‡,
- Millard H. Lambert¶,
- Kenneth H. Pearce‡,
- Thomas B. Stanley‡,
- Eugene L. Stewart¶,
- Ryan P. Trump**,
- Timothy M. Willson** and
- Shawn P. Williams¶
+ Author Affiliations
- ↵§ To whom correspondence should be addressed: Dept. of Gene Expression and Protein Biochemistry, GlaxoSmithKline, Five Moore Dr., Research Triangle Park, NC 27709. Tel.: 919-483-3821; Fax: 919-483-0368; E-mail; randy.k.bledsoe@gsk.com.
Abstract
Ligand binding is the first step in hormone regulation of mineralocorticoid receptor (MR) activity. Here, we report multiple crystal structures of MR (NR3C2) bound to both agonist and antagonists. These structures combined with mutagenesis studies reveal that maximal receptor activation involves an intricate ligand-mediated hydrogen bond network with Asn770 which serves dual roles: stabilization of the loop preceding the C-terminal activation function-2 helix and direct contact with the hormone ligand. In addition, most activating ligands hydrogen bond to Thr945 on helix 10. Structural characterization of the naturally occurring S810L mutant explains how stabilization of a helix 3/helix 5 interaction can circumvent the requirement for this hydrogen bond network. Taken together, these results explain the potency of MR activation by aldosterone, the weak activation induced by progesterone and the antihypertensive agent spironolactone, and the binding selectivity of cortisol over cortisone.
The mineralocorticoid receptor (MR)1 (NR3C2) is a steroid hormone-regulated transcription factor that plays a physiological role in the regulation of water and electrolyte (primarily sodium and potassium) levels in mammals. Although its effects in the distal nephron of the kidney and colon are thought to have the largest influence on vascular blood pressure, MR is found in many tissues, including the brain and heart (1-4).
MR is comprised of 784 amino acids and is the longest member of the oxosteroid receptor subgroup of the nuclear receptor (NR) superfamily, which includes the androgen receptor (AR), glucocorticoid receptor (GR), and progesterone receptors (PR) (5, 6). Amino acid alignments show that these receptors are very similar and contain three major functional domains: an N-terminal activation domain, a central DNA binding domain, and a C-terminal ligand binding domain (LBD) (7). Besides containing all of the determinants for binding hormone, the LBD also contains the C-terminal activation function-2 helix (AF-2). Crystal structures of several NR LBDs have shown that correct positioning of the AF-2 is required for recruitment of coactivators of transcription (8-10). Understanding the ligand requirements that lead to the proper positioning of the AF-2 and activation of MR is fundamental to designing drugs that can modulate receptor activation.
Within the body, MR is exposed to many steroids including cortisol, cortisone, and progesterone; however aldosterone and deoxycorticosterone (DOC) are considered the physiological MR ligands (Fig. 1). Circulating levels of the glucocorticoid cortisol, a potent MR binder and activator, may reach 1,000-fold over aldosterone (7, 11, 12). In epithelial tissues, high levels of the enzyme 11β-hydroxysteroid dehydrogenase type 2 convert cortisol to cortisone by converting the C-11 hydroxyl to the corresponding ketone (13, 14). This simple conversion results in a steroid incapable of activating MR. In a similar manner, progesterone levels are elevated over those of native MR ligands, especially during pregnancy (15, 16). Although there appear to be mechanisms in place for the conversion of this steroid to other metabolites, progesterone is normally a weak activator of MR even though it is known to bind with high affinity (17-20). This is particularly interesting because DOC, which differs from progesterone only at the C-21 position, is a potent activator of MR (11, 12).
Besides a role in hypertension, MR activation in cardiac tissue has recently been shown to play a role in cardiac fibrosis (21, 22). The synthetic steroid spironolactone (Fig. 1) has been used as an antihypertensive agent for decades and has recently received renewed interest as a treatment for congestive heart failure (23-25). Spironolactone is characterized by a C-17 γ-lactone ring and a C-7 thioester, but the molecular mechanism for the antagonist activity of the steroid has not been determined. Although crystallographic studies of other steroid hormone receptors have demonstrated the key role of the C-3 keto group in anchoring the steroid in the receptor binding pocket, the roles of other substituents of steroidal ligands have not been as clearly defined (9, 10, 26-28). A crystal structure of MR bound to an agonist or antagonist would therefore be useful in understanding the ligand requirements for modulation of MR activity.
In this report, we describe the crystal structures of MR bound to aldosterone, DOC, and progesterone. In addition, we describe the structure of the MR S810L mutant bound to progesterone, cortisone, and spironolactone. These structural studies, along with functional data from mutants, have revealed ligand requirements for MR activation and provide a rationale for the antagonistic activity of progesterone and spironolactone.
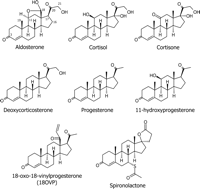
Structures of various steroids. The steroids aldosterone, cortisol, DOC, and 11-OH-P are agonists of MR (7, 11, 48). Cortisone binds the wtMR with very low affinity (30). Progesterone binds MR with high affinity but is a poor activator of the receptor (11, 31). The progesterone derivative 18-OVP is able to bind and activate MR (36, 48). Spironolactone is an antagonist of wtMR but is a potent agonist of MR containing the S810L mutation (31).
EXPERIMENTAL PROCEDURES
Construct Design and Protein Expression of Human MR LBD—A PCR was used to amplify the DNA encoding amino acids 712-984 of MR (GenBank M16801). The forward oligonucleotide contained a BamHI site, and the reverse oligonucleotide contained a HindIII site, which allowed for directional cloning of the amplified product into the modified His6-GST pET-24 vector (Novagen) digested with the same enzymes and described previously for the expression of GR (9). For the C808S and C808S/S810L mutants, two complementary oligonucleotides for each desired mutation were constructed: forward primer (C808S), 5′-GTA TTC TTG GAT GTC TCT ATC ATC ATT TGC CT-3′; reverse primer (C808S), 5′-AGG CAA ATG ATG ATA GAG ACA TCC AAG AAT AC-3′; and forward primer (C808S/S810L), 5′-TCT TGG ATG TCT CTA TTA TCA TTT GCC T-3′; reverse primer (C808S/S810L), 5′-AGG CAA ATG ATA ATA GAG ACA TCC AAG A-3′.
The underlined bolded letters depict the base changes from the wild type human MR sequence. The MR LBD (amino acids 712-984) cloned into the modified His6-GST pET-24 vector was used as the backbone to create the mutants. Sequences of the final constructs were confirmed. A thrombin protease site at the C terminus of the GST allows for cleavage of the resultant fusion protein following expression.
For protein expression, BL21(DE3) cells (Novagen/Invitrogen) were transformed with the expression plasmids. Expression was performed similar to that described previously for GR (9). Ligands (10-100 μm final concentration) were added 30 min prior to induction of expression. Proteins were purified to homogeneity using an AKTApurifier 100 (Amersham Biosciences) in a manner similar to that described for AR and PR (26-28).
Crystallization Methods—The MR LBD-hormone complex was concentrated to ∼4 mg/ml in buffer containing 25 mm HEPES pH 7.0, ∼190 mm NaCl, 10% glycerol, 10 mm dithiothreitol, 0.5 mm EDTA, 0.05% β-octyl glucoside. Crystals were grown at room temperature in hanging drops containing 3.0 μl of the above protein buffer solution and 1-5 μl of well buffer (0.8-1.0 m lithium sulfate, 0.1 m HEPES pH 7.5, 2% polyethylene glycol 2000 monomethyl ether). Crystals appeared over-night and grew continuously to 300 μm within a week. Diffracting crystals of MR were also obtained in the same conditions when micro-seeded with crushed MR·DOC crystals that were streaked with a horse tail hair. Before data collection, crystals were exchanged stepwise into the well buffer with an additional 25% ethylene glycol and were flash frozen in liquid nitrogen.
Diffraction data were collected on either a MSC/Rigaku RU200 generator with a MAR345 Image Plate detector or at the Advanced Photon Source IMCA 17 ID beam line on an ADSC 210 detector. Regardless of the hardware, diffraction data were integrated and scaled using HKL2000. All crystals were in two related crystal forms with either the space group C2221 or P212121, depending on whether the noncrystallographic dimer relating two subunits became a crystallographic 2-fold.
The first MR structure was solved by molecular replacement with the PR (PDB 1A28) using AMoRe. Several rounds of rebuilding with Quanta and refinement using CNX2002 resulted in the final models shown. All subsequent MR structures were obtained by molecular replacement with a refined MR model. All data collection and structure refinement parameters are included as supplemental data.
Cell Culture—CV-1 cells were maintained for growth in Dulbecco's modified Eagle' medium (Invitrogen) supplemented with 10% fetal bovine serum (Irvine Scientific, Santa Ana, CA), 100 units/ml penicillin G sodium, 100 μg/ml streptomycin sulfate (Invitrogen), and 2 mm l-glutamine (Invitrogen). Passaging was performed every 3-4 days at a 1:10 dilution. For experiments, CV-1 cells were plated in 96-well plates at a density of 20,000 cells/well in phenol red-free Dulbecco's modified Eagle's medium:nutrient mixture F-12 (Ham's) 1:1 (Invitrogen) supplemented with 10% charcoal/dextran-treated fetal bovine serum (Hyclone, Logan, UT), 100 units/ml penicillin G sodium, 100 μg/ml streptomycin sulfate, and 2 mm l-glutamine.
Cell Transfection and Ligand Treatments—Expression plasmids were made by cloning the sequence encoding the full-length MR into the pcDNA3 expression vector (Invitrogen). MR mutations at amino acid 770 or amino acid 945 were made by designing overlapping primers containing the mutation of interest. The oligonucleotides were then used in a PCR with the wtMR pcDNA3 plasmid as a template. Resultant PCR products were digested with DpnI and used to transform DH5α maximum efficiency competent cells to isolate clones of interest. The full MR and promoter region sequence was verified in all mutants. Transfections were performed 24 h after cells were plated. Lipofectamine reagent (Invitrogen) was used for transfecting cells and performed essentially according to the manufacturer's instructions. For cell-based reporter assays 10 ng of murine mammary tumor virus luciferase reporter, 4 ng of Renilla luciferase, 5 ng of MR expression plasmids, and varying amounts of pBluescript carrier plasmid were transfected into each well. Steroid dilutions were prepared in phenol red-free Dulbecco's modified Eagle's medium:nutrient mixture F-12 (Ham's) 1:1 supplemented with 100 units/ml penicillin G sodium, 100 μg/ml streptomycin sulfate, and 2 mm l-glutamine. Treatments included aldosterone, cortisol, DOC, progesterone, 11-hydroxyprogesterone (11-OH-P), and spironolactone (all at 1 μm). Cells were incubated for 24 h in the presence of the steroids, after which the medium was assayed for luciferase activity. Luciferase reporter activity was measured using the Dual Glo assay system (Promega).
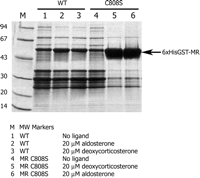
Effect of the MR C808S mutation on expression. Shown are the soluble Ni2+ resin-binding fractions. Lane 1, His6-GST-MR (wild type) expressed in the absence of ligand; lane 2, in the presence of 20 μm aldosterone; and lane 3, in the presence of 20 μm DOC. Lane 4, expression of MR C808S in absence of ligand; lane 5, C808S mutation in the presence of 20 μm aldosterone; and lane 6, C808S mutation in the presence of 20 μm DOC. As shown, the C808S mutation in combination with ligand allows for increased expression of soluble MR protein.
RESULTS
MR Interactions with Potent Agonists—Previous work on the steroid receptors PR, AR, and GR has demonstrated that receptor expression in Escherichia coli can be accomplished in the presence of high affinity ligands (9, 10, 26-28). In addition, expression studies with GR demonstrated that a single mutation at position 602 (F602S) led to increased expression and crystallization (9). When the equivalent MR residue, cysteine 808, was mutated to a serine, a dramatic increase in soluble expression of a GST MR LBD in E. coli was observed (Fig. 2). The MR C808S mutant yielded significant levels of protein when expressed in the presence of the potent agonists aldosterone and DOC. After expression, the protein was purified and crystallized as described.
Both MR-agonist complexes crystallized in the space group C2221 with one molecule in the asymmetric unit (see supplemental data). Overall, the MR LBD shows the three-layered α-helical fold observed in other nuclear receptor LBDs, with aldosterone bound in a fully enclosed pocket contacting residues in helices 3, 4, 5, 6, 7, and 11, and the β-turn (Fig. 3A). As seen in other steroid receptor LBDs, the C-terminal extension of MR interacts with helix 10 via hydrogen bonds between Asp929 and the amide nitrogens of Phe981 and His982. The most unusual feature of the structure is that the residues N-terminal to helix 1 (727-737) form a short helix that associates near the coactivator groove of a crystallographically related molecule (not shown). This N-terminal feature is present in all MR complexes.
In general, MR makes interactions with aldosterone in a manner consistent with how other steroid receptors bind their natural ligands (Fig. 3, B and C). There is an extensive hydrogen bond network involving the A-ring ketone of aldosterone, Gln776 and Arg817 of MR, and several water molecules that firmly lock the A-ring of the steroid in place. Specific to MR, there is a water-mediated hydrogen bond between Gln776 and Ser810. AR, GR, and PR have a methionine at this position so this interaction is not possible. Adjacent to the D-ring, the C-18 hydroxyl makes a hydrogen bond to the side chain carbonyl of Asn770 on helix 3. Asn770 is in position to coordinate a triplet of hydrogen bonds: one between the side chain carbonyl and the C-18 OH of the ligand, and two from the side chain nitrogen to the C-21 OH of the ligand and the backbone carbonyl of Glu955, a residue that lies on a loop preceding the AF-2 helix. The ligand is stabilized further by a pair of hydrogen bonds between the aldosterone C-21 hydroxyl, C-20 ketone, and Thr945 located on helix 10. This threonine is conserved in GR and PR but is replaced by leucine in AR. It is clear from the crystal structure that the orientation of Thr945 is ideal for hydrogen bonding with steroids containing C-20 carbonyls and C-21 hydroxyl groups (Fig. 3, B and C).
In the MR·DOC structure (not shown), only a single hydrogen bond between Asn770 and the C-21 OH exists. Additionally, Thr945 on helix 10 maintains a pair of hydrogen bonds to the C-20 ketone and C-21 OH of the steroid. Unique to aldosterone however, Cys942 appears in position to interact with the C-18 OH. This residue lies on helix 10 and thus provides aldosterone with three possible interactions with this helix.
In addition to Asn770, Ser767 is in position to make a hydrogen bond to the backbone amide of Glu955 in the loop preceding the AF-2. This observation suggests that both of the helix 3 residues (Asn770 and Ser767) play a role in stabilization and activation of MR. Amino acid alignments of the receptors show that an asparagine is conserved in a similar position in all of the oxosteroid receptors, suggesting that the interaction between helix 3 and the loop preceding the AF-2 may be a conserved mechanism of steroid receptor activation.
MR Interactions with Progesterone—Like DOC, progesterone has no substituents off the C-11 position of the steroid. However, DOC is a potent MR agonist, whereas progesterone has been characterized as a poor activator of MR (11). Using the MR C808S mutant to obtain the structure of the progesterone complex, the structural basis for decreased activation of MR by this ligand was examined.
Although the MR-progesterone complex crystallized in a different space group (P212121) compared with bound agonists (C2221), the two space groups are related by a crystallographic 2-fold axis, and nearly identical unit cell parameters suggest that the two crystal forms are related. Not surprisingly, the two molecules in the asymmetric unit of the MR-progesterone complex are both similar to each other, and the MR·DOC complex. Comparison of the MR·DOC and MR-progesterone complexes shows that both the overall protein fold and residue conformations around the binding pocket are generally similar.
There are differences, however, between the MR·DOC and MR-progesterone complexes in the interaction of the protein with the D-ring substituents of the steroids (Fig. 3D). First, Asn770 makes no contact with progesterone. Second, the hydrogen bonding pattern to Thr945 is different. Instead of two hydrogen bonds between the ligand and Thr945 as seen with aldosterone and DOC, only a single hydrogen bond is formed with progesterone. Additionally, multiple orientations were observed for the Thr945 side chain, suggesting that the intermolecular hydrogen bond to progesterone is in competition with intramolecular hydrogen bonds to the backbone carbonyls of Phe941 and Cys942. This competition is not visible in either the aldosterone or DOC structures.
It is also apparent from the MR-progesterone structure that both Asn770 and Ser767 are in the same location observed in MR-aldosterone structure. Although these residues may be critical for maintaining receptor conformation and activation, they are clearly not the only factors necessary for MR activation.
Mutagenesis Analysis of Asn770 in MR—Previous mutagenesis studies with MR have shown the importance of Asn770 in receptor activation (29). However, only a couple of mutations were examined in these studies. The dual interaction of Asn770 with both the ligand and the loop preceding the AF-2 observed in the MR-aldosterone and MR·DOC crystal structures prompted a more thorough analysis of this residue. Using transient transfection studies and a ligand concentration of 1 μm, we analyzed MR activation in CV-1 cells induced by the C-21 hydroxylated steroids aldosterone, DOC, and cortisol. MR activation induced by spironolactone and the C-21 methyl steroids progesterone and 11-OH-P was also determined.
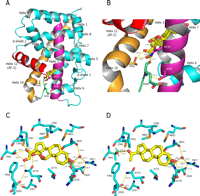
Crystal structure of MR LBD bound to aldosterone and progesterone. A, the overall fold of MR is very similar to the other steroid receptors. B, helix 3 (magenta) residues Asn770 and Ser767 form hydrogen bonds (yellow dashed lines) with the loop (green) residue Glu955 preceding the AF-2 (red). Thr945 present on helix 10 (orange) plays a key role in receptor activation by hydrogen bonding to the C-20 carbonyl and C-21 hydroxyl of aldosterone (yellow). C, close-up view of MR-aldosterone hydrogen bond network. The 18-OH is positioned for hydrogen bonding (yellow dashed lines) with the Asn770 carbonyl, whereas the Asn770 amide remains in position for hydrogen bonding to the C-21 OH of aldosterone and Glu955, which lies in the loop preceding the AF-2. Thr945, present on helix 10, forms a pair of hydrogen bonds with the C-20 and C-21 substituents of aldosterone. Cysteine 942 is in position to interact with the 18-OH group to give aldosterone three potential hydrogen bonds to helix 10. D, progesterone makes no hydrogen bonds to Asn770. Multiple orientations of the Thr945 side chain hydroxyl were observed (A and B) in both noncrystallographically related molecules. When the Thr945 side chain hydroxyl is in the B position (green), the distance (black dashed line) to the progesterone C-20 carbonyl makes hydrogen bonding between the ligand and Thr945 unlikely.
As expected, aldosterone, DOC, and cortisol showed potent activation of wtMR, whereas progesterone elicited a maximal response only ∼20% of the level achieved with aldosterone (Fig. 4). The progesterone derivative 11-OH-P, which differs from progesterone by the addition of a hydroxyl group at C-11, activated wtMR to ∼80% of the level achieved with aldosterone. Spironolactone failed to induce any appreciable MR activation at the 1 μm concentration. The mutations N770A, N770Q, N770S, N770D, N770T, and N770H all resulted in a reduction of activation to less than 5% of wt values with aldosterone, cortisol, DOC, or 11-OH-P (data not shown), whereas progesterone-induced activation was reduced to a lesser extent.
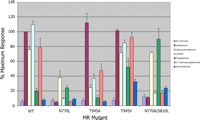
MR-mediated transactivation evaluated in the presence of various steroids. Wild type MR and mutants N770L, T945A, T945V, and S810L/N770A constructed in the expression vector pcDNA3 were transfected into CV-1 cells, and activation was evaluated in the presence of aldosterone, DOC, cortisol, progesterone, 11-hydroxyprogesterone, and spironolactone and compared with wtMR activation with aldosterone. All treatments were an n of 3. All steroids were tested at a concentration of 1 μm.
Interestingly, the N770L mutant was activated by DOC to ∼40% of wt levels, whereas progesterone-induced activation was unaffected (Fig. 4). These results suggest that a hydrophobic interaction between helix 3 and the loop preceding the AF-2 serves as a substitute for the hydrogen bonding normally observed with Asn770. The results also imply that any C-11 substituent on the steroid interferes with this new interaction. These results confirm the importance of Asn770 in the activation of MR and strengthen the assertion that interaction between helix 3 and the loop preceding the AF-2 is required for MR activation. Although it is possible that mutational analysis at this position could result in a receptor incapable of binding ligands, the N770A mutant has been shown to bind progesterone (but not aldosterone or cortisol) with nearly the same affinity as wtMR (29).
Mutational Analysis of Thr945 in MR—In the crystal structures, Thr945/ligand interactions appeared to distinguish potent agonists from the decreased activity observed with progesterone. To test the requirement for ligand/helix 10 interactions, the effects of mutating Thr945 on the activation of MR with both C-21-OH- and C-21-methyl-containing steroids were examined (Fig. 4). Aldosterone activation of the T945A mutant was equivalent to that of wtMR, whereas DOC and cortisol activation was less than 40% of the aldosterone-induced level. Progesterone and 11-OH-P activation was reduced to ∼10 and 50%, respectively, of the aldosterone maximum. Spironolactone failed to induce any activation of this mutant. An interaction of the ligand with the Thr945 side chain is clearly important for receptor activation.
The crystal structure of MR bound to progesterone suggests a molecular competition with respect to the orientation of Thr945. The interaction of progesterone with this residue fluctuates between a hydrogen bond with the C-20 carbonyl and the Thr945 hydroxyl, and a hydrophobic interaction between the C-21 methyl of the steroid and the Thr945 methyl. If this bifurcation occurs, mutation of Thr945 to a valine would remove the intramolecular hydrogen bond and possibly promote a stronger hydrophobic interaction with the C-21 methyl of progesterone derivatives, resulting in increased MR activation. Fig. 4 shows that the T945V mutation increased progesterone-induced MR activation from ∼20% to greater than 50% of the level of activation seen with aldosterone in wtMR. Activation of the T945V mutant by 11-OH-P was increased to a level nearly equivalent to that obtained by aldosterone, DOC, and cortisol with wtMR. In a similar manner, activation of the T945V mutant by the C-21-hydroxylated steroids aldosterone and DOC is unchanged relative to wtMR, although cortisol-induced activation is reduced slightly to ∼85% wtMR levels. Interestingly, spironolactone-induced activation increased to ∼30% of the level seen with aldosterone-induced wtMR, indicating that spironolactone is a partial agonist of this mutant. This assertion was strengthened further in experiments showing that 10 μm spironolactone was not capable of reducing aldosterone-induced activation of the T945V mutant to the levels achieved using wtMR (see supplemental data). These results suggest that although MR activation with C-21 OH steroids via hydrogen bonding with the Thr945-OH may be optimal, a substitute interaction between the ligand and helix 10, whether hydrophobic or via hydrogen bonding, can result in activation of MR.
A MR Mutant with Increased Ligand Binding and Transcriptional Activation—It has been shown that the S810L mutation in MR leads to potent activation by progesterone, spironolactone, and cortisone (30, 31). To determine the molecular basis for this increased activation, the MR C808S/S810L mutant was expressed and purified in the presence of these ligands, and the crystal structures were determined. The MR C808S/S810L progesterone complex crystallized in the same space group as the C808S mutant, facilitating a direct comparison of the protein conformation. The root mean square deviation between the two structures is only 0.20 Å, which is only slightly larger than the deviation between noncrystallographically related subunits (0.15 Å).
The conformations of residues within the binding pocket are nearly identical between the two forms of the protein (Fig. 5A). Because Thr945 again adopts two different orientations, this suggests that the increased activation of the MR C808S/S810L mutant in the presence of progesterone is not the result of a strengthened interaction with Thr945.
In the single mutant, Ser810 binds a water molecule that occupies a hydrophobic pocket adjacent to Ala773, Gln776, Met777, Trp806, Met807, and the ligand (Fig. 3, C and D). In the double mutant, the side chain of Leu810 occupies this pocket. The Leu810 side chain makes hydrophobic contacts with the C-19 methyl group of progesterone and with the Ala773 Cβ, as predicted earlier (31). Previous models of the MR S810L mutant have predicted that substitution of a leucine at position 810 would lead to steric hindrance between the leucine side chain and the side chain of Gln776 (32). However, this concept does not appear to be substantiated by the crystal structures.
Although it has been predicted that the S810L mutation leads to a bent helix 3, an overlay of the Cα-chains of progesterone bound to both the wild type and S810L mutant shows that the helix 3 position does not differ in the two structures. However, the crystal structures do reveal a slight kink in helix 5 around the S810L mutation that points toward helix 3, indicating that the helix 3/helix 5 interaction proposed by Geller et al. (31) may be partially correct.
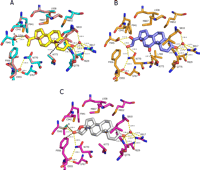
Close-up views of the MR C808S/S810L mutant bound to progesterone, cortisone, and spironolactone. A, an overlay of progesterone (yellow) in MR C808S (cyan) and progesterone (pink) in MR C808S/S810L (pink) binding pockets indicates that the S810L mutation has no measurable effect on ligand position or orientation. Hydrogen bonding networks (yellow dashed lines) are indicated. Van der Waals interactions are indicated by black dashed lines. Multiple orientations of the Thr945 side chain hydroxyl were observed (A and B) in both noncrystallographically related molecules. B, the C-11 carbonyl of cortisone (blue) does not interact with MR C808S/S810L (orange) residue Asn770 and is ∼2.9 Å away from the backbone carbonyl of Leu769. C, the lactone keto group of spironolactone (white) is ∼3.7 Å away from Asn770 in MR C808S/S810L (magenta), suggesting a weakened potential for hydrogen bonding to Asn770. Again, multiple orientations of the Thr945 side chain hydroxyl were observed (A and B) in both noncrystallographically related molecules. Similar to that observed with progesterone, the distance from the hydroxyl of Thr945 to the lactone keto moiety of spironolactone differs depending upon the orientation of this residue. The weak interaction of spironolactone with both Asn770 and Thr945 is probably the basis for the antagonism observed with this steroid.
Activation of the MR S810L Mutant—To determine the relative importance of the increased helix 3/helix 5 interaction in the S810L mutant and the hydrogen bonding to Asn770, the N770A mutation was introduced into the MR S810L mutant. The MR S810L/N770A mutant was not activated significantly by aldosterone. Cortisol- and 11-OH-P-induced activation was only ∼20% of the level achieved with aldosterone in wtMR. Spironolactone treatment resulted in partial activation (∼25%) of the mutant (Fig. 4). In contrast, DOC-induced activation levels were equivalent to those observed in wtMR. Interestingly, spironolactone was not able antagonize DOC-induced activation of the S810L/N770A mutant at a 10 μm concentration (see supplemental data), suggesting that spironolactone is an agonist (albeit with reduced affinity) of this mutant. Progesterone-induced activation was highest among the ligands tested with activation levels similar to those obtained with aldosterone in wtMR.
These results suggest several important considerations. First, proper orientation of Asn770 to preserve the helix 3/loop interaction is absolutely critical for activation with the C-21-OH steroids aldosterone and cortisol whether or not the S810L mutation is present. Second, because DOC and progesterone were capable of activating MR N770A/S810L whereas 11-OH-P was not, this suggests that polar substituents in the C-11 position of the steroid destabilize the helix 3/loop interaction in this mutant, resulting in decreased MR activation. These two considerations, combined with the fact that Asn770 must act as a hydrogen bond donor to the backbone carbonyl of Glu955 to stabilize this interaction, suggest that only C-21-OH ligands with hydrogen bond donors at the C-11 position will activate transcription. Steroids such as cortisone, in which a carbonyl at the C-11 position acts as a hydrogen bond acceptor, would not be expected to bind or activate MR. Thus, the molecular basis for selectivity of cortisol over cortisone by MR appears to be dictated by the absolute requirement for a specific orientation of Asn770. Interestingly, the S810L mutation allows cortisone to become a potent binder and activator of MR. In fact, hypertension in patients harboring the S810L mutation is probably because of MR being activated by the normally inactive cortisone (30). This consideration prompted us to obtain a crystal structure of the MR C808S/S810L mutant with cortisone.
Structure of MR C808S/S810L with Cortisone—In the MR/cortisone binding site, the hydrogen bonds between Asn770 and Ser767 to Glu955 are seen, along with hydrogen bonds from the C-20 carbonyl and C-21 OH to Thr945, thus satisfying the two requirements for receptor activation (Fig. 5B). As expected, the specific orientation of Asn770 prevents hydrogen bonding to the C-11 carbonyl of cortisone. Although it is possible that some electrostatic repulsion exists between the C-11 carbonyl and the backbone carbonyl of Leu769 in helix 3 (distance 2.9 Å), steric hindrance by the C-11 ketone of the steroid is not likely the only reason for the failure of cortisone to bind and activate wtMR. The structure does suggest interaction of Leu810 with the C-19 methyl of cortisone, and this is probably the basis for the increased affinity of cortisone for this receptor. It is not evident to what extent the increased ligand affinity or helix 3/helix 5 interaction combines to allow cortisone to activate this receptor.
Structure of MR C808S/S810L with the Antagonist Spironolactone—Expression of the MR C808S/S810L protein in the presence of the antihypertensive agent spironolactone also allowed for determination of this structure. Not surprisingly, spironolactone packed into the active site in the same manner as other steroid ligands (Fig. 5C). Spironolactone, like progesterone, makes a shorter hydrogen bond with Arg817 (2.8 Å) than with Gln776 (3.3 Å), in contrast to aldosterone, which makes 3.0 Å bonds to both residues. The lactone keto group of spironolactone is ∼3.7 Å away from Asn770, suggesting a weakened potential for hydrogen bonding to this residue. Likewise, the lactone keto moiety distance from the hydroxyl of Thr945 varies depending upon the orientation of the threonine side chain, suggesting a weakened hydrogen bonding capability to helix 10. The C-7 thioester does not appear to make any interactions with the protein. In fact, the ligand density ends after the sulfur atom, suggesting that the formyl group is rotating freely. Not surprisingly, spironolactone derivatives, such as canrenone, which are modified at this position, still retain activity (33, 34). Interestingly, there is no perceptible movement of the AF-2 helix in the MR-spironolactone complex. Spironolactone does not impinge on Leu960, the AF-2 residue that caps the binding pocket. The most significant contact observed is between the lactone ketone oxygen and the side chain of Phe956, which is just prior to the AF-2. Spironolactone is closer to this residue than any other steroid examined by ∼0.6 Å.
DISCUSSION
Mechanism of MR Activation—In this report, we present multiple crystal structures of MR complexed with steroidal ligands that modulate receptor activity differentially. Mutagenesis and activation studies combined with the structural information provide insight into the determinants of MR binding and activation by steroidal ligands. As summarized in Table I, steroidal activation of MR requires (in addition to the C-3 ketone) that the ligand forms hydrogen bonds to both Asn770 in helix 3 and Thr945 in helix 10. However, these requirements for activation can be overcome for the most part in the MR S810L mutant by a helix 3/helix 5 interaction. It is clear from the mutagenesis studies that the S810L mutation alone does not negate the need for Asn770 to stabilize the loop preceding the AF-2, especially in the presence of C-21 hydroxyl-containing steroids with C-11 substituents.
General observations derived from multiple MR crystal structures with various ligands
Stabilization of the loop preceding the AF-2 helix appears to be maximized when the amide side chain of Asn770 simultaneously acts as a hydrogen bond acceptor from the ligand and a hydrogen bond donor to the backbone carbonyl of Glu955. This dual interaction may act to polarize the side chain, reinforcing both of the individual hydrogen bonds. Notably, interference with the interaction between helix 3 and the loop preceding the AF-2 reduces MR activation (35). Two properties of steroid ligands dictate that an asparagine is required at position 770 in MR to stabilize the helix 3/loop interaction. First is the length of the steroid itself, and second are substituents at the C-11 position of the steroid. Neither aldosterone nor cortisol binds the MR N770A mutant, whereas progesterone binds with nearly the same affinity (29). Our own studies show that even the MR S810L/N770A double mutant has limited activation in the presence of the C-21 OH steroids aldosterone and cortisol. However, the S810L/N770A mutant is activated by DOC and progesterone, suggesting that the helix 3/helix 5 interaction induced by the S810L mutation can aid receptor activation (even in the absence of Asn770) in the presence of certain steroids (Fig. 4). One might expect that DOC would be capable of activating the S810L/N770A mutant to the same level as progesterone because this steroid contains no C-11 substituents. However, the C-21 hydroxyl of DOC occupies more volume than the progesterone C-21 methyl. This extension may strain the weak helix 3/loop interaction in the absence Asn770.
Ligand-induced stabilization of the loop preceding the AF-2 in MR appears especially critical because there are no direct hydrogen bonds between the AF-2 helix and helix 3 comparable with those observed in AR and PR (26-28) (Fig. 6, A and B). Unlike MR and GR, both AR and PR contain a glutamate on helix 3 which is capable of forming multiple hydrogen bonds directly with the end residue of the AF-2 helix, effectively locking the AF-2 in place. Because the crystal structure of GR reveals that the AF-2 helix is not stabilized via hydrogen bonds directly to helix 3 and because natural GR ligands are C-21 OH steroids, Asn564 may play a similar but less critical role in this receptor (9).
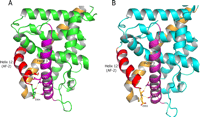
Stabilization of the AF-2 helix in PR and AR versus MR and GR. A, a glutamate (Glu723) present on helix 3 (magenta) in PR (green) stabilizes the AF-2 (red) by forming hydrogen bonds with the end residue of the AF-2 (28). Glu709 in AR performs a similar function (26, 27). In addition, PR residues Asn719 and Thr716 form hydrogen bonds to the backbone amide and carbonyl of Glu904. B, the lack of a direct interaction between helix 3 (magenta) and the AF-2 helix (red) in MR (cyan) and GR makes stabilization of helix 3/loop region (yellow) preceding the AF-2 helix more critical in these receptors. In MR, Asn770 and Ser767 form hydrogen bonds to the backbone of Glu955. This hydrogen bond arrangement dictates the orientation of Asn770 and positions the side chain to serve as a hydrogen bond acceptor to C-11 substituents on steroidal ligands. This explains why cortisone, which contains a hydrogen bond-accepting carbonyl at the C-11 position, is a low affinity MR ligand.
The MR crystal structures also reveal that a second residue on helix 3, Ser767, is in position to form a hydrogen bond with the amide portion of the Glu955 backbone. This interaction probably adds additional stability to the helix 3/loop interaction in MR. Interestingly, a S767Q mutation caused complete loss of activation with steroidal ligands, and a S767N mutation yielded greatly reduced receptor activity (data not shown). Therefore, it appears that the hydrogen bond formed by Ser767 is necessary but not independently sufficient for stabilization of the loop preceding the AF-2 helix.
The combined data also reveal the importance of residue Thr945. The crystal structures of MR with progesterone show that the orientation of the Thr945 side chain is ambiguous because of competition between the ligand and intramolecular hydrogen bond acceptors (Figs. 3D and 5A). This reduction in hydrogen bonds (and hydrogen bond strength) combined with no interaction with Asn770 is most likely the molecular basis for the weak activation seen with progesterone.
In our studies, replacement of Thr945 with an alanine led to decreased MR activation with DOC, cortisol, and 11-OH-P but not with aldosterone (Fig. 4). Aldosterone and cortisol binding affinities for MR T945A have been reported to be similar (29). Thus, differences in cortisol- and aldosterone-induced activation levels suggest that aldosterone makes an additional interaction with helix 10 through another substituent other than the C-21 hydroxyl.
Activation levels of the MR T945V mutant induced by the C-21 OH-containing steroids aldosterone and DOC were equivalent to the levels observed with wtMR. However, activation induced by the C-21 methyl steroids progesterone and 11-OH-P was increased. Again, strengthening of the interaction between the ligand and helix 10 leads to increased activation of MR. This does not rule out the possibility that an interaction between the ligand and a residue other than Thr945 on helix 10 would also lead to receptor activation.
It is possible that aldosterone contributes a third hydrogen bond to helix 10 through the 18-OH of the steroid and Cys942. This interaction would explain why aldosterone is able to activate the T945A mutant to a greater extent than either DOC or cortisol (100% versus less than 40%). The progesterone derivative 18-oxo-18-vinylprogesterone (18-OVP) (Fig. 1) may be another ligand that activates MR by this mechanism. Even in the context of a N770A mutation, MR activation has been observed in the presence of 18-OVP, suggesting that the C-18 oxygen of the ligand is critical for activation through interaction with another residue other than Asn770 (36, 37). As predicted, and based on the MR crystal structures, the most plausible explanation for activation by this compound is an interaction between Cys942 and the C-18 enone oxygen providing two possible interactions of the ligand with helix 10 (37). Whether the interaction with Cys942 also results in an increased hydrophobic interaction between the C-21 methyl group of 18-OVP and the Thr945 methyl or reduces the hydrogen bonding distance between the Thr945 OH and the carbonyl of the ligand, MR activation is achieved by an increased interaction of the ligand with helix 10.
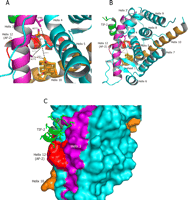
Stabilization of helix 3 (magenta) in the MR S810L mutant (cyan). A, close-up view of van der Waals interactions (black dashed lines) of Leu810 with the C-19 methyl of spironolactone and the side chain carbons of Ala773, Met777, and Val780. B, ribbon view of MR S810L bound to spironolactone. A short fragment of the TIF-2 cofactor (green) is modeled into the docking site for the LXXLL motif of the peptide. C, surface view of the same complex. Stabilization of helix 3 (magenta) must occur for proper docking of the cofactor peptide (green).
Mechanism of Activation in the MR S810L Mutant—An S810L mutation in MR has been reported to yield a receptor capable of being activated with progesterone, spironolactone, and cortisone (30, 31). Combining the C808S mutation with the S810L mutation allowed us to obtain crystal structures. A comparison of the single and double mutant MR-progesterone structures reveals that the only difference seen is residue 810 (Fig. 5A). When the MR S810L mutation was first described, it was predicted that a hydrophobic interaction between the Leu810 side chain and the Cβ of Ala773 caused a bending of helix 3, which led to receptor activation (31). The MR S810L progesterone structure supports some hydrophobic interaction between these two residues as they are ∼4.2 Å apart. However, the crystal structures suggest that Leu810 is also in van der Waals contact with two other residues on helix 3. The side chain sulfur atom and a side chain carbon atom of Met777 are 3.7 and 4.0 Å, respectively, from the leucine side chain, and the side chain of Val780 is 4 Å from the Leu810 side chain (Fig. 7A). That Leu810 is making multiple contacts with helix 3 is supported by the fact that an A773G mutation in the context of the S810L mutant leads only to a modest decrease in norprogesterone-induced MR activation (31).
There is precedence in the steroid receptors that a helix 3/helix 5 interaction aids ligand-induced receptor activation. Similar to that seen previously in AR, GR, and PR, the steroidal ligand in MR is anchored in the binding pocket of the LBD by the polar residues Gln776 in helix 3 and Arg817 in helix 5 (9, 10, 26-28). Thus, the C-3 keto group of the steroid is responsible for bridging a helix 3/helix 5 interaction already. The S810L mutation enhances this interaction further and reduces the requirement for ligand-mediated hydrogen bonding to Asn770 and Thr945. However, it is clear that even the MR S810L mutant requires stabilization of the loop preceding the AF-2 because the C-21 OH-containing steroids aldosterone and cortisol were not able to activate this receptor (in the context of the N770A mutation) to a significant degree (Fig. 4).
Structurally, it is evident why the helix 3/helix 5 interaction is important for receptor activation. Cofactors such as steroid receptor coactivator-1 and transcriptional intermediary factor-2 (TIF-2) play a key role in steroid receptor-mediated transcription. These proteins contain a signature LXXLL motif (or NR box) and are known to interact with the steroid LBD through the docking of this motif into a hydrophobic grove formed by helices 3, 4, and AF-2 (9, 10, 38). Together the steroid receptor and cofactor protein form a part of the complex needed for gene transcription.
Fig. 7, B and C, shows a superposition of the MR C808S/S810L spironolactone structure with a peptide representing the third LXXLL motif of TIF-2 (39). It is clear that proper positioning of both helix 3 and the AF-2 must be achieved for the LXXLL-containing transcription factors to bind. The S810L mutation may aid stabilization of helix 3 in this conformation via van der Waals interactions with Ala773, Met777, and Val780. Because the AF-2 helix must also be in proper position for binding of the α-helical LXXLL sequence motifs found in many coactivators, the MR S810L mutant still requires ligand for maximal activation. However, the interaction of a ligand (like progesterone) with helix 10 would likely be more important than any interaction with Asn770.
Antagonism of MR and MR S810L—Spironolactone is an antihypertensive that has been used clinically for several decades. The crystal structures of MR C808S/S810L with spironolactone and progesterone provide insights into the requirements needed for receptor activation and also provide insights into the molecular basis of MR modulation. MR antagonism by progesterone and spironolactone is a “passive” antagonism. These ligands bind and prevent MR from adopting the active conformation by failing to mediate hydrogen bonding to Asn770 and Thr945. The result is that both helix 3 and the AF-2 helix are not arranged in the proper position to allow efficient binding of coactivators of transcription.
This mode of antagonism differs from the “active” antagonism seen when a ligand such as RU486 binds GR or when raloxifene binds to the estrogen receptor. In these examples, the dimethylaniline group of RU486 and the pyridine of raloxifene physically prevent the respective receptors AF-2 from adopting the active conformation (10, 40). Although the methyl side chain of Ala773 prevents wtMR from binding RU486, ligands that physically destabilize the interaction between Asn770 and the loop preceding the AF-2 could result in a similar antagonism (11, 41). Likewise ligands that impinge on helix 10 or the AF-2 will likely result in receptor antagonism. Finally, because of the apparent importance of the helix 3/helix 5 interaction in receptor activation in MR as well as GR and PR, it has been suggested that ligands that interfere with this interaction would be potential antagonists (42).
Recently, ligands capable of antagonizing MR containing the S810L mutation have been reported (32). Based on our results, it is likely that these ligands fail to activate MR S810L for a variety of reasons. 5α-pregnan-20-one does not contain a C-3 carbonyl group. Although the S810L mutation aids stabilization of helix 3, loss of interaction with Gln776 (helix 3) could negate any benefit of the mutation, especially since this steroid has no substituent present to interact with Asn770. The second steroid that inactivates MR S810L is 4,9-androstadiene-3,17-dione. This steroid is characterized by a C-17 carbonyl that is unlikely to be in position to hydrogen bond with helix 10. Finally, RU486 can bind the MR S810L mutant; however, the dimethyl aniline will physically prevent the mutant from obtaining the active conformation of the AF-2 as well as destroying the helix 3 interaction with the loop preceding the AF-2, thus resulting in an inactive receptor.
Role of Mutations in the MR LBD in Mineralocorticoid Resistance—Mineralocorticoid resistance or type 1 pseudohypoaldosteronism is a rare condition characterized by neonatal renal salt wasting and a failure to thrive. The autosomal dominant and sporadic forms of the disease are caused by mutations in the MR. To date three missense mutations, Q776R, L924P, and L979P in the MR LBD, have been linked with this disease (43-46). Determination of the MR crystal structures allows for pinpointing the location and probable consequences of each of these mutations.
The MR crystal structures confirm that residue Gln776 (helix 3) plays an important role in anchoring the C-3 keto group of the steroid in the binding pocket. Mutation of this residue to an arginine results in a receptor with a diminished affinity for aldosterone and reduced transactivation in transient transfection assays (46). It is also possible that this mutation prevents the correct positioning of helix-3 required for efficient binding of LXXLL-containing cofactors. Based on the MR crystal structures, residue Leu924 lies in the third turn of helix 10 facing the interior of the protein. Incorporation of a proline at this point would serve to break this important helix, resulting in an altered protein conformation incapable of steroid binding. Finally, Leu979 lies in the β-sheet immediately following the AF-2 helix. As predicted, and verified by the MR crystal structures, the backbone amide of Leu979 is in position to hydrogen bond with the backbone carbonyl of Ser879, which lies on the opposite strand of the β-sheet (47). Mutation of Leu979 to a proline is thought to destabilize this β-sheet resulting in the lack of aldosterone binding and transactivation observed with this mutant (46, 47).
CONCLUSION
We have shown that maximum MR activation occurs only when there is simultaneous stabilization of the loop preceding the AF-2 helix and a strong interaction of the ligand with helix 10. Stabilization of the loop preceding the AF-2 requires hydrogen bonds between Asn770 and Ser767 on helix 3 and Glu955 present on this loop. Ligands that promote this hydrogen bond network and interact with helix 10 via hydrogen bonds or hydrophobic interaction with Thr945 induce a stabilization of helix 3 and a movement of the AF-2, enabling coactivator recruitment and ultimately gene transcription. This series of ligand-mediated activation steps ensures that ligands such as progesterone and cortisone fail to activate MR even though these ligands will be in excess over aldosterone in many tissues. Likewise, spironolactone also fails to activate MR because of an inability to create the hydrogen bonding network and thus behaves as a passive MR antagonist.
The mutation S810L in MR has two consequences. First, the receptor has an increased affinity for ligands through interaction of Leu810 with the C-19 methyl of the steroid (30, 31). The second consequence is an induced stabilization of helix 3 through van der Waals interactions with multiple residues on helix 3. This induced stabilization of helix 3 in the absence of ligand reduces but does not abolish the need for interaction of ligand with helix 3 and helix 10 for MR activation to occur. Together the multiple MR crystal structures provide insights into the requirements for receptor activation and will aid in the quest for novel ligands that effectively modulate MR activity resulting in the discovery of improved drugs for the treatment of hypertension and heart failure.
Acknowledgments
We thank all of our colleagues in our respective departments for helpful insights and discussions. We especially thank Thomas Consler and John Gray for support with this project.
Footnotes
-
↵1 The abbreviations used are: MR, mineralocorticoid receptor; AF-2, activation function-2; AR, androgen receptor; DOC, deoxycorticosterone; GR, glucocorticoid receptor; GST, glutathione S-transferase; His6, hexahistidine; LBD, ligand binding domain; NR, nuclear receptor; 11-OH-P, 11-hydroxyprogesterone; 18-OVP, 18-oxo-18-vinylprogesterone; PR, progesterone receptor; TIF-2, transcriptional intermediary factor-2; wt, wild type.
-
The atomic coordinates and structure factors (codes 2AA2, 2AA7, 2AA5, 2AA6, 2AAX, and 2AB2) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
-
↵* The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
-
↵[boxs] The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1 and 2 and Fig. 1.
- Received April 14, 2005.
- Revision received June 8, 2005.
- The American Society for Biochemistry and Molecular Biology, Inc.













