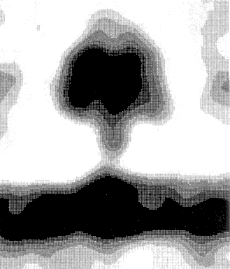
 This image is of the complete complex, using image averaging and cryo-electron microscopy,
from Rod Capaldi's homepage.
This image is of the complete complex, using image averaging and cryo-electron microscopy,
from Rod Capaldi's homepage.
Lecture 10 ATP synthase |
The ATP synthase enzymes have been remarkably conserved through evolution. The
bacterial enzymes are essentially the same in structure and function as those from
mitochondria of animals, plants and fungi, and the chloroplasts of plants. The early
ancestory of the enzyme is seen in the fact that the Archaea have an enzyme which is
clearly closely related, but has significant differences from the Eubacterial branch. The
H+-ATP-ase found in vacuoles of the eukaryote cell cytoplasm is similar to the
archaeal enzyme, and is thought to reflect the origin from an archaeal ancestor.
In most systems, the ATP synthase sits in the membrane (the "coupling"
membrane), and catalyses the synthesis of ATP from ADP and phosphate driven by a flux of
protons across the membrane down the proton gradient generated by electron transfer. The
flux goes from the protochemically positive (P) side (high proton electrochemical
potential) to the protochemically negative (N) side. The reaction catalyzed by ATP
synthase is fully reversible, so ATP hydrolysis generates a proton gradient by a reversal
of this flux. In some bacteria, the main function is to operate in the ATP hydrolysis
direction, using ATP generated by fermentative metabolism to provide a proton gradient to
drive substrate accumulation, and maintain ionic balance.
In mitochondria, the P side is the intermembrane space, and the N side the mitochondrial
matrix; in bacteria, the P side is the outside (the periplasm in gram negative bacteria),
the N side the cytoplasm; in chloroplasts, the P side is the lumen and the N side the
stroma.
There are minor differences between bacteria, mitochondria and chloroplasts in some of
the smaller subunits, which leads to a confusing nomenclature. The simplest system is that
from E. coli. The ATP synthase can be dissociated into two fractions by relatively
mild salt treatments.
A soluble portion, the F1 ATP-ase, contains 5 subunits, in a
stoichiometry of 3a:3b:1g:1d:1e. Three
substrate binding sites are in the b-subunits. Additional
adenine nucleotide binding site in the a-subunits are
regulatory. The F1 portion catalyzes ATP hydrolysis, but not ATP-synthesis.
Dissociation of the the F1 ATP-ase from the membranes of bacteria or organelles
leaves behind a membrane embedded portion called FO. This consists (in E.
coli) of three subunits a, b and c, with relative stoichiometries of 1:2:9-12. The
c-subunit is very hydrophobic, and forms a helix turn helix structure which spans the
membrane twice, with a hydrophilic loop on the side of attachment of F1. There
is a conserved acidic residue half-way across the membrane in the C-terminal helix.
After dissociation, the membranes are permeable to protons. The proton leak can be stopped
by addition of inhibitors, which are also inhibitors of ATP synthesis in the functional
complex. Two "classical" inhibtors are commonly used. Oligomycin binds at the
interface between Fo and F1; dicyclohexylcarbodiimide (DCCD) binds
covalently to the conserved acidic residue in the c-subunit of Fo. One DCCD per
ATP-ase is sufficient to block turn-over, suggesting a cooperative mechanism. The action
of these inhibitors indicates that the proton permeability of the Fo is a part
of its functional mechanism.
The proton leak can be plugged, and a functional ATP synthase can be reconstituted, by
adding back the F1 portion to membranes containing the Fo portion.
This image is of the complete complex, using image averaging and cryo-electron microscopy,
from Rod Capaldi's homepage.
The structure of the soluble (F1) portion of the ATP synthase from beef
heart mitochondria has been solved by X-ray crystallography. The pictures below are from
Abrahams, J.P., Leslie, A.G., Lutter, R. and Walker, J.E. (1994)
Structure at 2.8 Ĺ resolution of F1-ATPase from bovine heart mitochondria.
Nature 370, 621-628.
The protein was crystallized in the presence of ADP, and an ATP analogue, AMP-PNP, in
which the the two terminal phosphates of ATP were replaced by the non-hydrolysible
imidodiphosphate group. The three a-subunits each contained an
AMP-PNP. The three b-subunits contained either ADP (bDP), AMP-PNP (bTP),
or no nucleotide (bE).
Click on images for large version.
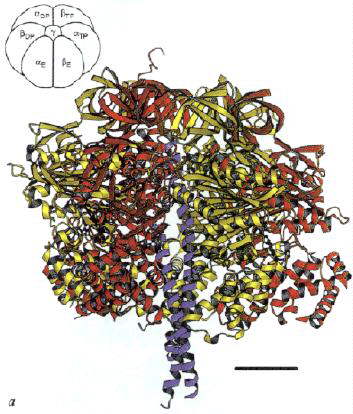
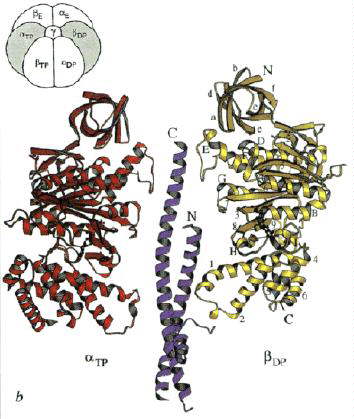
Left: The structure of the F1 ATP-ase, viewed from the side. The a-subunits are shown in yellow, theb-subunits
in red, and the g-subunit in blue. The cartoon at the top left
shows the orientation. Note that the a- b-subunits
alternate in a ring around the g-subunit, which forms a rod up
the middle. The a- and b-subunits
are differentiated by subscript indicating the occupancy of the active site of the b-subunit of each a- b-pair:
E - empty; DP - ADP; TP - ATP analogue, AMP-PNP. Scale bar is 20 Ĺ.
Right: A vertical slice through the complex across the a-TP/b-DP diagonal highlighted in the cartoon. 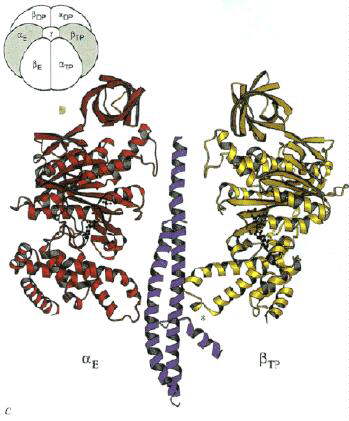
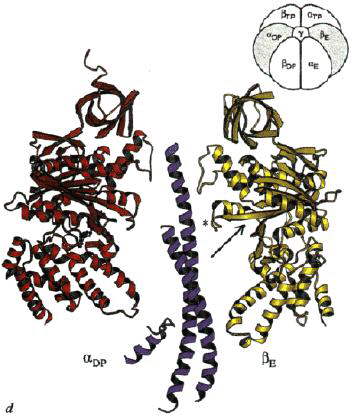
Left: A vertical slice through the complex across the a-E/b-TP diagonal highlighted in the cartoon.
Right: A vertical slice through the complex across the a-DP/b-E diagonal highlighted in the cartoon.
Note how the "jaw" of the nutcracker swings open when the site is empty
(arrow in picture on right). 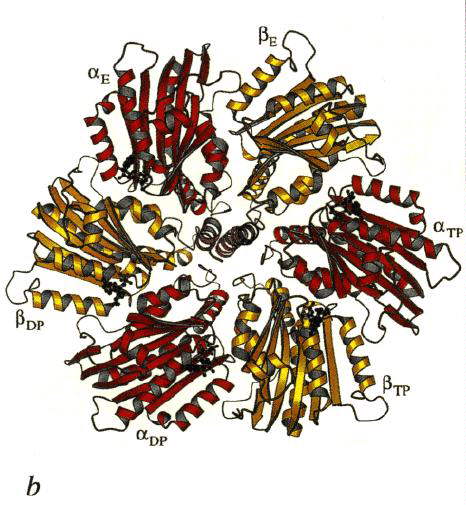
View of the complex from the top (N-phase). 

Left: A horizontal slice through the complex across the top, showing the b-sheet structure which provides a cap over the catalytic domain.
Scale bar is 20 Ĺ.
Right: A horizontal slice through the complex across the catalytic domain, which is
predominantly helical. 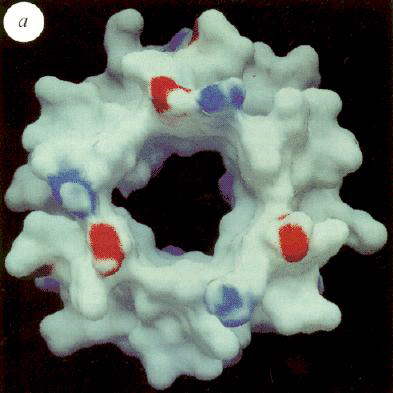

Left: View of the calculated electrostatic surface potential of the a,
b-sleeve formed by the structure below the b-sheet
cap, showing regions of negative (red) and positive charge, and a predominantly neutral
(hydrophobic) "hole" in the dough-nut, through which the top part of the g-subunit protrudes. The view is from inside the protein.
Right: Similar surface, but viewed from the side, of the g-subunit,
showing a hydrophobic surface for most of the rod, but a marked neatively charged polar
region half-way down. The upper part of the rod slides into the sleeve shown in the Fig.
to the left, as indicated in cross-section by the ball-and-stick structure. 
Cross-section through the structure showing surfaces, and highlighting the fit of the g-subunit in the a, b-ring.
Also shown is the location of the bound ATP analogue (AMP-PNP) in the bTP-subunit.
Note how the bulge, introduced in the g-subunit by the
horizontal helix, abutts against the bTP-subunit,
and forces a change in conformation. It is suggested that rotation of the g-subunit in the a, b-ring
induces conformational changes in successive a, b-pairs so as to bring about the binding changes expected from the
binding-change mechnism (see below).
The ATP synthase operates through a mechanism in which the three active sites undergo a
change in binding affinity for the reactants of the ATP-ase reaction, ATP, ADP and
phosphate, as originally predicted by Paul Boyer. The change in
affinity accompanies a change in the position of the g-subunit
relative to the a, b-ring, which
involves a rotation of the one relative to the other. In the direction of ATP synthesis,
the rotation is driven by a flux of H+ down the proton gradient, through a
coupling between the g-subunit, and the c-subunit of FO.
This rotation has now been demonstrated experimentally.
Click here for some nice animation movies of the F1
ATPase mechanism in action, by Hongyun Wang and George Oster, University of
California, Berkeley.
Two of these movies are available locally.
A perspective view of a3,
b3, g in cartoon display
with stereo on.
A top view of a3, b3, g in cartoon display.
This rotational motion has been captured in dramatic
videos from the laboratory of Masasuke Yoshida. In this work, the F1-ATPase
was tethered to a glass surface by the b-subunit, using a
His-tag engineered into the protein at the N-terminus, and NTA-ligand on the glass (see
illustration from Junge et al. TIBS article, below). 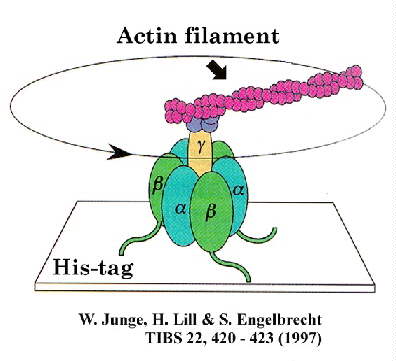
The motion was detected by attaching an actin filament to the g-subunit,
which was tagged with fluorescent groups to make it visible, and recorded using a video camera attached to a microscope. The motion was seen
only under conditions of ATP-hydrolysis, and the direction of motion was always
counter-clockwise when viewed from the Fo portion, giving the sign of the
catalytic mechanism.
Hiroyuki Noji, Ryohei Yasuda, Masasuke Yoshida & Kazuhiko Kinosita Jr. (1997) Direct
observation of the rotation of F1-ATPase. Nature, 386, 299 - 302.
An alternative approach using photometric methods has been explored in Wolfgang Junge's
lab. Use of small chromophores attached directly to the g-subunit
has the advantage of a time resolution unconstrained by the large torque associated with
the movement of the actin filament above. Two methods have been used to explore the
dynamics of the system. 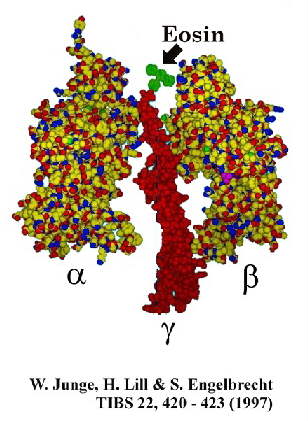
In [1], the authors used photoselection experiments with a small chromophore (eg. eosin,
see above) attached to the g-subunit of the active F1-ATPase,
and observed the relaxation of polarization anisotropy on activation of turnover. The
behavior was compatible with that expected for a three-stepped rotatory device. In [2],
they extended their kinetic analysis of the rotation, aided by a new theory for assessing
continuous versus stepped, and Brownian versus unidirectional molecular motion. The
observed relaxation of the absorption anisotropy was fully compatible with a
unidirectional and stepping rotation of g over three
equidistantly spaced angular positions in the hexagon formed by the alternating subunits a and b. The results strongly supported a
rotational catalysis with equal participation of all three catalytic sites.
In [3], polarized confocal fluorometry (POCOF) was applied to single molecules of
engineered, immobilized and load-free spinach-CF1 ATP-ase, and was used to
investigate transition states of the rotatory drive. Hydrolysis of ATP caused the stepped
and sequential progression of subunit g through three discrete
angular positions, with the transition states of g being too
shortlived for detection. The authors also observed the stepped motion of e, whereas d, a
and b subunits were immobile.
Reference [4] is a brief and readable review of this work.
Siggi Engelbrecht's homepage
for some nice images, and for pdb files of homology models for spinach F1-Atp-ase (with models of d and e subunits, and E. coli F1, F0-ATPase (with models of d, e subunits, and c and b subunits from F0).(The pictures below are from Duncan, T.M., Bulygin, V.V., Zhou, Y.,
Hutcheon, M.L. and Cross, R.L. (1995) Rotation of subunits during catalysis by E. coli
F1-ATPase. Proc. Natl. Acad. Sci., USA 92, 10964-10968.) 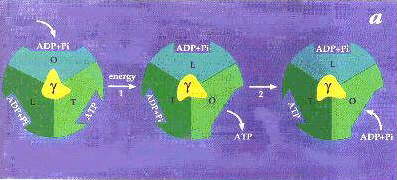 In the above cartoon showing the
binding-change mechanism of Paul Boyer, rotation of the g-subunit
(yellow) relative to the a, b-ring
(the three a, b-pairs are
represented by different shades of green or blue) induces a change in the binding
affinities of reactants, as represented here by a change in the conformation of the site
on going from left to right in the diagram. In step 2, ATP forms spontaneously from
tightly bound ADP and Pi. The mechanism was proposed before the structure was known, so
the structure provides a nice confirmation of the model. The Open site corresponds
to the Empty site of the structure, the Tight site to the ATP site,
and the Loose site to the ADP site.
In the above cartoon showing the
binding-change mechanism of Paul Boyer, rotation of the g-subunit
(yellow) relative to the a, b-ring
(the three a, b-pairs are
represented by different shades of green or blue) induces a change in the binding
affinities of reactants, as represented here by a change in the conformation of the site
on going from left to right in the diagram. In step 2, ATP forms spontaneously from
tightly bound ADP and Pi. The mechanism was proposed before the structure was known, so
the structure provides a nice confirmation of the model. The Open site corresponds
to the Empty site of the structure, the Tight site to the ATP site,
and the Loose site to the ADP site.
Experimental evidence for the model comes from an extensive history of research:
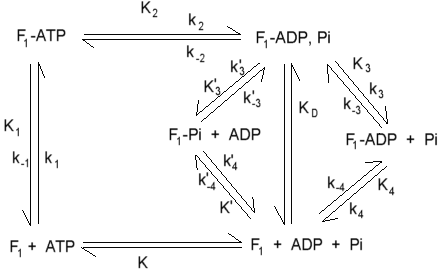
Equilibrium (K) and kinetic (k) constants for hydrolysis of ATP by F1 under uni-site turnover conditions. Values for some of the constants are:
k1 = 6.4 x 106 M-1sec-1
k-1 = 7 x 10-6 sec-1
K1 = ~1012 M-1
k2 = 12 sec-1
K2 = 0.5
k3 = 2.7 x 10-3 sec-1
k4 = 3.6 x 10-4 sec-1
k-4 = 1.3 x 103 M-1sec-1
K4 = 0.3 x 10-6 M
K'4 = 80 x 10-6 M
K = 3.6 x 105 M
Since the structure has become available, a lot of nice work has been done to test the rotational model discussed above. Among the most convincing experiments are those from the paper above, which are shown schematically in the figure below. 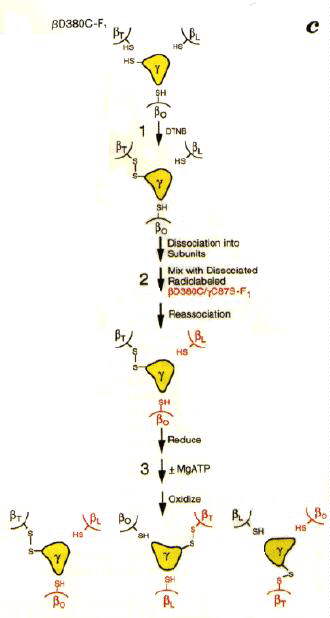
The experiments depend on the fact that cysteine can often be substituted into a protein in place of other amino acids in the sequence without the mutation effecting the function. When two cysteines are close enough together in a structure, addition of an oxidant (Aox below) will induce formation of a disulphide (R-S-S-R) cystine bond between them. The bond can be broken, and the cysteines reformed, by addition of a reducing agent.
Aox + 2 R-SH <==> AH2 + R-S-S-R In addition, the F1 could be reversibly dissociated into subunits without breaking the disulphide bond, so that the b-subunit could be removed and added back.
In these experiments, a cysteine introduced into the g-subunit (C87S) and a cysteine introduced in place of D380 of the b-subunit by site-directed mutations (D380C), were used to set up the system so that a cross-bridge could be formed between g and b-subunits. Since there is only one g per complex, only one of the three potential cross-bridges was formed in each F1. It had previously been shown that formation of the cross-bridge inactivated the enzyme. Side by side experiments were done with unlabelled and 35S-isotopically labelled F1-ATP-ase.
When the unbridged and bridged subunits were separated, it was found that new bridges had been formed between unlabelled b-subunits, and labelled g-subunits in the ratio expected for a rotational mechanism. Control experiments ± ATP, Mg2+, etc., showed that the rotation indicated by the transfer of the disulphide bond required turnover of the enzyme. 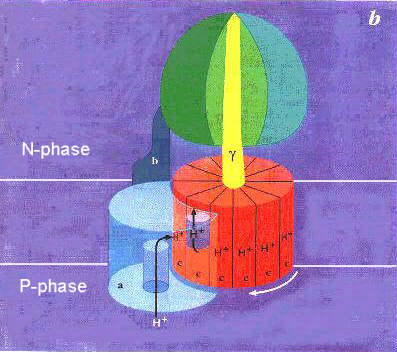
|Cartoon showing the two parts of the ATP synthase, with a rotation of g-subunit driven by coupling to a "motor" consisting of the c-subunits of FO. The c-subunits form a complex which moves in the membrane with respect to the a-subunit of FO. The idea suggested by Wolgang Junge (click here to see a model ) is that the a-subunit provides a port for entry of protons from the P-phase, and a port for exit to the N-phase. When a proton enters through the P-phase port, it neutralizes the conserved acidic residue in the helical hairpin of the c-subunit. Only in this neutral form (animation from Hongyun Wang's Home Page) can the c-subunit rotate away from association with the a-subunit. Rotation brings a neutral c-subunit to the exit port, allowing it to lose the proton, and associate with the a-subunit complex. Successive protonations allow the c-subunit complex to rotate by 1/n x 360o for each proton, where n is the stoichiometry of the c-subunit per ATP synthase (9-12). Because a complete rotation drives ATP synthesis at each of the 3 catalytic sites, 3 or 4 H+ are required for each ATP,- the stoichiometry found. DCCD (see above) blocks the mechanism by acting as a covalent "spanner", jamming the works when bound to any single c-subunit.
Click here for an animation of the complete mechanism.
Experiments from Capaldi's lab, using engineered placement of cysteine residues to explore the neighborliness of subunits through formation of disulphide bridges, suggest that the b-subunits, together with the d-subunit of F1 form a stator, attached near the "top" of a b-subunit, which prevents the a, b-ring from moving. The e-subunit can be attached to g-, c-, a- or b-subunits. Presumably it changes its attachment to the a, b-ring, in order to allow the rotation with respect to g which is now an established part of the mechanism. 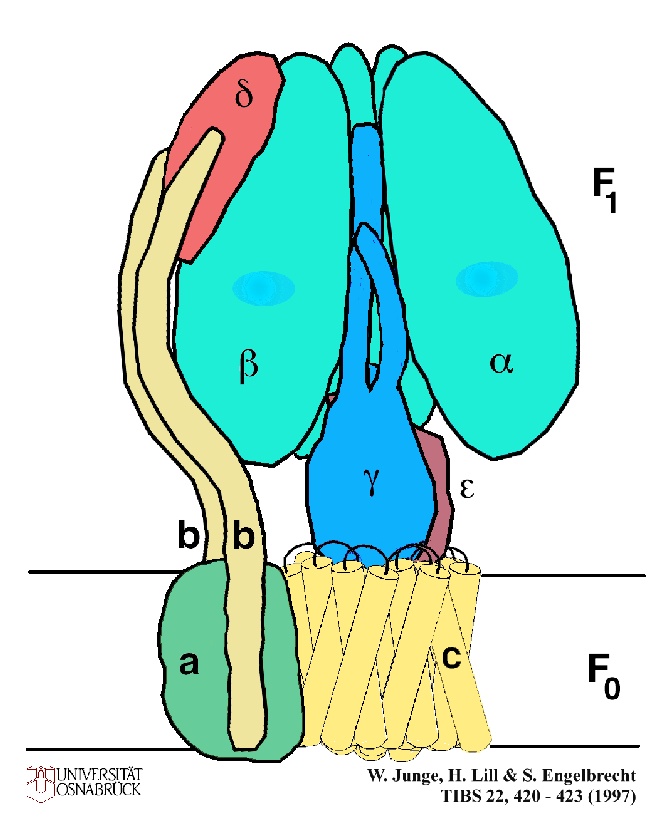
A cartoon showing the arrangement of the subunits which join the F1 section to the FO section. Note that the stalk of the ATP synthase has now become two stalks, one central, composed of the e- and g-subunits, linked to the c-subunit complex, and the other peripheral, composed of the d- and b-subunits. Why is this second stalk not seen in electron microscopy images? Capaldi suggests that the reason reflects the averaging which is necessary to get high quality images. Symetrical structures like the a, b-ring and the central stalk will contribute to the average, but asymetric structures like the peripheral stalk will be "averaged out" of the image, unless special care is taken to select images with such a feature in a fixed orientation. ![]()
{kind=link}
{kind=link}