
The Productive Conformation of Arachidonic Acid Bound to Prostaglandin Synthase
Back to the Index...COX+ Author Affiliations
Prostaglandin H synthase-1 and -2 (PGHS-1 and -2) catalyze the committed step in prostaglandin synthesis and are targets for nonsteroidal anti-inflammatory drugs (NSAIDs) like aspirin. We have determined the structure of PGHS-1 at 3 angstrom resolution with arachidonic acid (AA) bound in a chemically productive conformation. The fatty acid adopts an extended L-shaped conformation that positions the 13proS hydrogen of AA for abstraction by tyrosine-385, the likely radical donor. A space also exists for oxygen addition on the antarafacial surface of the carbon in the 11-position (C-11). While this conformation allows endoperoxide formation between C-11 and C-9, it also implies that a subsequent conformational rearrangement must occur to allow formation of the C-8/C-12 bond and to position C-15 for attack by a second molecule of oxygen.
PGHSs convert AA, O2, and two electrons to prostaglandin H2 (PGH2) in the committed step in prostaglandin synthesis. PGHSs catalyze two semi-independent reactions (1,2)—a bis oxygenase [cyclooxygenase (COX)] reaction that uses AA and two O2molecules to form PGG2 and a peroxidase (POX) reaction in which PGG2 undergoes a two-electron reduction to PGH2. Two isoforms of PGHS (PGHS-1 and -2) are found in most, if not all, mammalian tissues. PGHS-1 is constitutively expressed and is involved in prostaglandin biosynthesis in response to hormone stimulation. The resulting prostanoids interact with cell surface G protein (heterotrimeric GTP–binding protein)–linked receptors to mediate “housekeeping” functions, including the regulation of renal water and sodium metabolism, stomach acid secretion, parturition, and hemostasis. In contrast, PGHS-2 is an inducible enzyme that is expressed transiently in response to growth factors, tumor promoters, or cytokines. Prostanoids produced via PGHS-2 are involved in cell replication and differentiation. Both PGHS-1 and -2 are of pharmacological importance because they are targets for aspirin and NSAIDs. Aspirin inhibition of PGHS-1 lowers the risk for mortality from cardiovascular disease (3, 4), whereas inhibition of PGHS-2 acts to reduce inflammation, fever, and pain (5–7); various cancers (8–12); and possibly Alzheimer's disease (13). The recently approved COX-2 inhibitors, which lack the gastrointestinal side effects associated with more classical NSAIDs (14, 15), directly target PGHS-2.
The crystal structures of ovine PGHS-1 (oPGHS-1) (16–18) and murine and human PGHS-2 (19, 20) have elucidated many of the interactions that drugs make with these enzymes. However, the productive modes of binding for AA and other fatty acid substrates within the COX active site have only been inferred (1, 2, 21). The formation of stable complexes of native PGHS with AA has been difficult, as trace amounts of contaminating peroxides and hydroperoxides lead to the generation of tyrosyl and arachidonyl radicals, product formation, and, ultimately, enzyme inactivation (22). To circumvent this problem, we reconstituted apo-oPGHS-1 with Co3+-protoporphyrin IX to create a native-like enzyme species (Co3+-oPGHS-1) that lacks both POX and COX activities and does not form any prostaglandin products when incubated with AA (23). Moreover, the co-crystallization of Co3+-oPGHS-1 and fatty acid substrates occurs without the formation of oxidation products in the crystal (23). Here, we present the x-ray crystal structure of Co3+-oPGHS-1 complexed with the fatty acid substrate AA.
The hexagonal crystals of the Co3+-oPGHS-1:AA complex can be flash-frozen for data collection at low temperature (23). The data from two separate crystals were used for two independent structure determinations, one (AA-1) at 3.0 Å resolution and the other (AA-2) at 3.1 Å (24) (Table 1). This strategy was chosen because there is substantial variation in the diffraction quality of oPGHS-1 crystals (25). Because both structures were identical within experimental error (Table 1), we discuss only the interpretation of the AA-1 structure. The NH2-terminal epidermal growth factor (EGF) domain, the membrane binding domain (MBD), and the catalytic domain (Fig. 1) are well resolved in the Co3+-oPGHS-1:AA complex. Moreover, the Co3+-protoporphyrin IX, the sugars at the three N-linked glycosylation sites, and four detergent molecules bound to the MBD are also well resolved. Bound AA was clearly observed in the initial 2F o − F c(F o, observed structure factor;F c, calculated structure factor) electron density maps. Iterative model building, with the use of standardF o − F cdifference electron density maps and simulated annealing omit maps followed by positional refinement, allowed the accurate placement of AA in the active site (Fig. 2) despite the limited resolution.
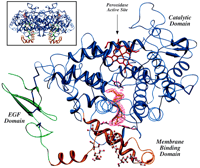
A ribbon representation of the Co3+-oPGHS-1 monomer with AA bound in the COX channel. The EGF domain, MBD, and catalytic domain are shown in green, orange, and blue, respectively; Co3+-protoporphyrin IX is depicted in red, disulfide bonds (Cys36-Cys47, Cys37-Cys159, Cys41-Cys57, Cys59-Cys69, and Cys569-Cys575) in dark blue, and side chain atoms for COX channel residues Arg120, Tyr355, and Tyr385 in magenta. Also shown is a F o −F c simulated annealing omit map contoured at 1.5σ for AA (yellow). The four β-OG detergent molecules (ball and stick) are shown bound to the MBD, including one detergent molecule found just below Arg120 and Tyr355 within the opening to the COX channel. (Inset) The native dimer with the twofold axis running vertical. All figures were created fully or in part using SETOR (44).
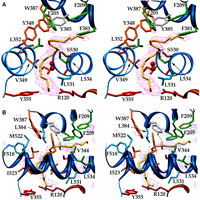
AA bound in the COX channel (30). (A) Stereo view of AA bound in the COX channel. TheF o − F csimulated annealing omit electron density contoured at 1.5σ for AA (yellow) is shown. Side chain atoms for all residues that contact the substrate at the carboxylate, C-2 through C-11, and C-14 through C-20 are colored red, orange, and green, respectively. Ser530 (magenta), which is acetylated by aspirin, lies below Tyr385 (gray), the likely radical donor during catalysis. Leu531 (light blue) lies above Arg120 but does not contact the substrate. The two red spheres represent the presumed location of O2 for attack on C-11. (B) The view of the COX active site rotated 90° about the vertical axis [using the same color scheme as in (A)]. TheF o − F cdifference density (purple) around the COX molecule is contoured at 2.5σ; the largest peak in the map constitutes that for AA. Residues Phe518, Leu384, and Met522 (in light blue) along with Phe381 and Trp387 constitute the endoperoxide pocket. Hydrogen atoms for C-13 have been modeled. The 13proS hydrogen is 2.3 Å from the OH group of Tyr385.
Summary of data collection and refinement statistics.
AA is bound within the COX active site and adopts an extended L-shaped conformation with two kinks in the center (Fig. 2). Carbons of AA C-1 through C-3 bind in the channel with the carboxylate positioned to interact with the guanidinium group of Arg120 and the phenolic oxygen of Tyr355. C-7 through C-14 form an S shape that weaves the substrate around the side chain of Ser530. C-13 is close to the phenolic oxygen of Tyr385 and is oriented properly for abstraction of the proS hydrogen. The S shape also positions C-11 above a small pocket into which O2 could presumably migrate from the lipid bilayer (Fig. 2A). Thus, C-11 would be accessible to O2 from the side opposite (antarafacial) to hydrogen abstraction, a known aspect of the reaction (26). The ω-end of the substrate (C-14 through C-20) binds in a cul-de-sac along helices 6 (residues 325 through 353) and 17 (residues 520 through 535) between Ser530 and Gly533 (Fig. 2).
As expected, the residues lining the COX channel make multiple hydrophobic interactions with AA (Fig. 3). Of 49 interactions between the enzyme and substrate, only the two carboxylate interactions with Arg120 and Tyr355 are hydrophilic. Fatty acid–enzyme contacts can be divided into three spatial regions in terms of the substrate carbons: the apical end (C-1 through C-7), the “catalytic” core (C-8 through C-13), and the ω-end (C-14 through C-20). Some of the more interesting substrate-protein interactions are discussed below.
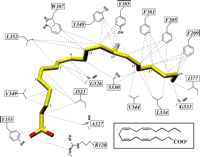
A schematic of interactions between AA and COX channel residues (30). Carbon atoms of AA are yellow, oxygen atoms red, and the 13proS hydrogen blue. All dashed lines represent interactions within 4.0 Å between the specific side chain atom of the protein and AA; the structures AA-1 and AA-2 revealed the same set of 49 contacts. Only two of these contacts between AA and the COX channel residues are hydrophilic. The carboxylate forms a salt bridge to the guanidinium atom of Arg120 (distance = 2.4 Å; angle = 143°) and a hydrogen bond to the OH group of Tyr355 (distance = 3.1 Å; angle = 115°). (Inset) A schematic of the chemical structure of AA.
At the apical end, the carboxylate of AA forms an edge-on salt bridge with the guanidinium group of Arg120 (Fig. 2B). Arg120 lies at the base of the COX active center above the MBD, protruding into the COX channel. Earlier studies (16–18) predicted that the interaction of the AA carboxylate with Arg120 was a major determinant in substrate binding. Indeed, the mutation of Arg120 to glutamine increases the K m (Michaelis constant) of oPGHS-1 for AA by 1000-fold, demonstrating how critical this ionic interaction is for substrate binding to PGHS-1 (27). In contrast, Arg120 in PGHS-2 is not critical for substrate binding (28), a result that cannot yet be adequately explained.
Tyr355 lies across from Arg120 at the base of the COX channel (Fig. 2) such that its hydroxyl group forms a hydrogen bond to the carboxylate of AA. This interaction appears important in the discrimination between the stereoisomers of certain NSAIDs and may also play a role in substrate binding (27). Leu531 is also in close proximity to Arg120, and its Cδ1 methyl group abuts the carboxylate of AA (Fig. 2A). Mutating Leu531 to lysine inactivates the enzyme (29), most likely due to competition with Arg120 for the substrate's carboxylate. Similarly, the Leu531 → Asp531 (L531D) (30) and L531N mutations have native-like K m values but only one-tenth of the native COX activity (29). Apparently, the introduction of hydrophilic residues at residue 531 can lead to stable but improper liganding of the substrate carboxylate to the enzyme. Thus, the proper AA binding to Arg120is critical for enzyme activity in PGHS-1, and even modest structural changes in the immediate environment are not tolerated.
Above the carboxylate end of AA, the fatty acid chain runs along Ile523 and then bends over Val349, Ala527, and Ser530 (Fig. 2). Ile523 makes hydrophobic contacts with C-2 and the C-5/C-6 double bond (Fig. 3). Residue 523, which is a valine in PGHS-2, is the only residue in the first shell of COX active site residues that differs between PGHS-1 and PGHS-2. Having a valine at this position in PGHS-2 increases the size of the COX channel, a feature that is exploited by PGHS-2–specific NSAIDs (31) and may account for some isozyme differences in substrate utilization (32). However, the Cγ2 atom of Ile523 abuts the substrate, a contact that could also be present with Val523 in PGHS-2. The I523V (30) mutation in PGHS-1 decreases the COX activity by 30% (33), a result that demonstrates that subtle—and perhaps more global—differences exist between nearly identical active sites in the two isozymes.
Residues in the catalytic core region surround the center of the substrate (C-8 to C-13) and the radical donor/acceptor Tyr385. These residues can be divided into classes: those that come within 4 Å of the substrate (Tyr348, Leu352, Trp387, and Ser530) and those that do not (Leu384, Phe518, and Met522). Ser530, which is acetylated by aspirin treatment, abuts against the substrate (Fig. 2A); its Cα and Cβ atoms make hydrophobic contacts with C-10 and C-16, while the Oγ atom lies underneath C-13. When Ser530 is replaced with alanine, the enzyme retains 60 to 80% of its native COX activity without changing theK m for AA (34). Thus, the serine hydroxyl is neither essential for catalysis nor for AA binding. A S530T (30) mutation reduces the COX activity to 15% that of native enzyme, with a sixfold increase inK m (29,34). Modeling an extra methyl group at this position suggests that the mutation affects the conformation of the neighboring residues (Gly526, Ala527, and Leu534), which contact both the carboxyl and ω ends of the substrate.
Phe381, Leu384, Trp387, Phe518, and Met522 form a pocket that could accommodate the conformational transitions involving C-8 through C-12 during O2 addition and endoperoxide ring formation (Fig. 2B). Of these residues, Leu384, Phe518, and Met522 do not contact the substrate, whereas Trp387 contacts both the substrate and Tyr385, the putative radical donor. C-11, the first site of attack by O2, and C-12 abut against the ring edge of Trp387. The recently constructed W387F and W387L (30) mutants (33) produce considerable amounts of 11(R)-hydroperoxy-eicosatetraenoic (11R-HETE), presumably arising from the failure to form the endoperoxide bridge. Thus, Trp387 may help properly align or stabilize intermediates during PGG2 formation.
Positioned below Tyr385, the phenolic group of Tyr348 hydrogen bonds to the phenolic group of Tyr385, an interaction seen in all other PGHS-1 crystal structures (16–18). A Y348F (30) mutation has no effect on COX activity (35), indicating that the conserved hydrogen bond between the hydroxyl groups of Tyr348 and Tyr385 is not important for catalysis. However, Tyr348 makes three hydrophobic contacts with carbons 12 through 14 of the substrate( Fig. 3 ) and may help position C-13 for hydrogen abstraction.
The ω-end of AA lies between helices 6 and 17 such that Phe205, Phe209, Leu344, Phe381, and Leu534 form a hydrophobic cage around the last six carbons of the substrate (Figs. 2 and 3). At C-17, the end of AA turns abruptly, placing C-20 against Gly533. The G533A (30) mutant in PGHS-1 has no COX activity (29), suggesting that a clash between methyl group of alanine and C-20 is enough to prevent the proper binding of AA. The hypothesis that the more spacious COX active site in PGHS-2 affords added variability in substrate binding (20, 32,36) is supported by the fact that the G533A (30) mutant in PGHS-2 retains some COX activity (37). Only when Gly533 in PGHS-2 was mutated to a leucine or valine was all COX activity lost (37). The larger COX active site in PGHS-2 is also used to explain why aspirin acetylation inactivates COX activity in PGHS-1 but allows turnover of AA to 15R-HETE in PGHS-2 (38). The recent work by Rowlinson et al.(37) shows how the mutation of Gly533 in PGHS-2 alters the enzyme's activity toward 18 carbon unsaturated fatty acids, suggesting that the shape and size of this binding region may be important in determining substrate selectivity.
The observed structure of AA bound in the COX active site agrees well, on a qualitative level, with the productive conformations of AA proposed by a recent modeling study (37) and a crystal structure of mouse PGHS-2 containing a mixture of product and substrate (21). It also places considerable constraints on any proposed mechanism for enzyme catalysis. However, it must be noted that PGHS-1 and -2 do not convert AA to PGG2 with 100% efficiency (26). The PGHS isoforms make low levels of 11R-HETE and 15(R/S)-hydroperoxyeicosatetraenoic acids (15-HETE) due to the existence of minor alternate conformers of AA within the COX active site (26). Accordingly, the observed electron density for AA in oPGHS-1 may be an average of more than one AA conformer. Nonetheless, the modeled conformation of AA satisfies the stereochemical requirements of the initial catalytic steps, allowing a structurally valid sequence of catalytic events for the COX reaction to be envisioned (Fig. 4).
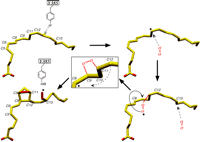
Mechanistic sequence for converting AA to PGG2 (30). Abstraction of the 13-proS hydrogen by the tyrosyl radical leads to the migration of the radical to C-11 on AA. Attack of molecular oxygen, coming from the base of the COX channel, occurs on the side antarafacial to hydrogen abstraction. As the 11R-peroxyl radical swings over C-8 for an R-side attack on C-9 to form the endoperoxide bridge, C-12 is brought closer to C-8 via rotation about the C-10/C-11 bond allowing the formation of the cyclopentane ring. The movement of C-12 also positions C-15 optimally for addition of a second molecule of oxygen, formation of PGG2, and the migration of the radical back to Tyr385.
AA is positioned such that a Tyr385 phenolic radical can abstract the 13proS hydrogen (Fig. 2B). Oxygen then migrates into a small pocket (Val349, Ala527, Ser530, and Leu531) (Fig. 2A) below C-11 and attacks the substrate from the side antarafacial to that of hydrogen abstraction to yield an 11R-peroxyl radical intermediate (26). The 11R-peroxyl radical then attacks C-9 to produce the endoperoxide bridge, which produces a radical centered on C-8 (Fig. 4). However, the extended conformation of AA cannot permit facile ring closure between C-12 and C-8 at this stage of the reaction because the distance between C-8 and C-12 is about 5 Å. Thus, a major reconfiguration of the substrate must occur concomitant with or immediately following formation of the endoperoxide bridge.
We propose that the conformational transition involves substantial movement of the ω-end of AA toward the apical half of the molecule. The loss of the C-11/C-12 double bond with the formation of the 11R-peroxyl moiety increases the local conformational freedom for the attack on C-9. The peroxyl could then swing “over” C-8 for a R-side attack on C-9 (Fig. 4), bringing C-12 closer to C-8 via rotation about the C-10/C-11 bond. This conformational transition would also reposition atoms C-13 to C-20. AS-side attack on C-9 is less likely due to steric hindrance from C-7. Formation of the endoperoxide bridge also results in the loss of the C-8/C-9 double bond, which again increases local conformational freedom. The radical, now on C-8, is ready for attack on C-12 (Fig. 4). Additionally, these conformational transitions in the substrate could also position C-15 optimally for the addition of the second O2 molecule and for hydrogen donation by Tyr385 to the 15S-peroxyl radical. The latter event completes the COX reaction and returns the radical to the catalytic tyrosine for the next turnover.
Further mutagenic and structural studies of oPGHS-1 interactions with fatty acids are needed to confirm the catalytic events proposed here. The functional differences between PGHS-1 and PGHS-2 in response to identical mutations (e.g., R120Q and G533A) (30) also remain unexplained, but suggest that the two isoforms have subtle but distinct differences in active site structures, which remain to be elucidated.
-
↵* To whom correspondence should be addressed. E-mail: garavito@magaera.bch.msu.edu
- Received for publication 17 February 2000.
- Accepted for publication 30 June 2000.


