

A Novel Family of Hydroxamate-Based Acylating Inhibitors of Cyclooxygenase
Back to the Index...COX+ Author Affiliations
- Desmond Fitzgerald, Dept. of Clinical Pharmacology, Royal College of Surgeons in Ireland, 123 St. Stephens Green, Dublin 2, Ireland. E-mail dfitzgerald@rcsi.ie
Abstract
Aspirin irreversibly inhibits cyclooxygenase (COX) by acetylating a serine residue in the active site. We synthesized a series of novel acylating agents based on our previously reported acetylating compound,O-acetylsalicylhydroxamic acid. One of these, triacetylsalicylhydroxamic acid (TriAcSHA) was more effective than aspirin and O-acetylsalicylhydroxamic acid in inactivating both COX-1 and COX-2. Preincubation of COX-1 with inhibitor for 5 min yielded IC50 values of 18 μM for TriAcSHA and 60 μM for acetylsalicylic acid. Inhibition was time-dependent, with complete inhibition within 10 min at a concentration of 50 μM. As with aspirin, mutation of the serine 530 of COX-1 to alanine abolished the activity of the TriAcSHA. Mutation of the alanine 119 to a glutamine markedly reduced the sensitivity to TriAcSHA, suggesting that this residue was necessary for the interaction with the enzyme. TriAcSHA was also more effective than aspirin as an inhibitor of platelet aggregation induced by arachidonic acid. The diacetylated phenylhydroxamatesN-methyl-O,O-diacetylsalicylhydroxamic acid, N,O-diacetylbenzohydroxamic acid, and 2-methyl-O,N-diacetylbenzohydroxamic acid showed reduced or absent activity against COX-1. In addition, we synthesized a series of triacylsalicylhydroxamic acids with progressively longer acyl groups (three to six carbons). All of the compounds inhibited COX-1 and demonstrated progressively greater COX-1 selectivity with increasing number of carbons. Hence, salicylhydroxamic acid provides a versatile backbone for the generation of a family of acylating inhibitors of cyclooxygenase.
Cyclooxygenase (COX) is the enzyme responsible for the generation of prostaglandins, bioactive lipids that are involved in many pathophysiological processes including vascular thrombosis (FitzGerald et al., 2000), temperature control, and nocioception (Aronoff and Neilson, 2001). Two isoforms of the enzyme have been identified that are the products of distinct genes (Appleby et al., 1994). COX-1 is expressed ubiquitously and is the only form in platelets, where it generates thromboxane, a potent platelet activator. COX-2 is rarely found in normal tissue but is expressed at sites of inflammation, in tumors, and in response to tissue injury. Both isoforms of COX are inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs) (Marnett et al., 1999), which are widely used in the management of pain and arthritis. Aspirin [acetylsalicylic acid (ASA)] is unique among the NSAIDs in that it covalently modifies the enzyme. Aspirin acetylates a serine residue in the substrate channel of the enzyme, inactivating COX-1 and converting COX-2 to a 15-lipoxygenase and preventing prostaglandin generation (Loll et al., 1995).
Despite the apparent simplicity and usefulness of aspirin, few acetylating compounds have been generated. Those that have depend on the delivery of an acyl group from the phenolic oxygen. Attempts to generate aspirin-like drugs with increased PGH2synthase isoform specificity have resulted in acetoxybenzene compounds with O-alkynylether or thioether substituents (Kalgutkar et al., 1998). These compounds inactivate both COX-1 and -2; in some cases, however, they show selectivity for isoform-2. In contrast, valerylsalicylic acid was found to be moderately selective for COX-1 (Bhattacharyya et al., 1995). We recently described in this journal a novel acetylating inhibitor,O-acetylsalicylhydroxamic acid (AcSHA), in which the acetyl group was delivered from a hydroxamic acid backbone rather than the phenolic group (Loll et al., 2001). We have used this structure to generate a series of acylating compounds, including triacetylsalicylhydroxamicacid (TriAcSHA), an acetylating agent derived from the salicylhydroxamic acid scaffold that is more potent and structurally very different from aspirin.
Experimental Procedures
Materials.
Dulbecco's modified Eagle's medium, fetal bovine serum, penicillin, phosphate-buffered saline, RPMI 1640 medium, streptomycin, and trypsin were obtained from Invitrogen (Paisley, UK). COX-1 and arachidonic acid were purchased from Cayman Chemical Company (Ann Arbor, MI). Tris, hematin,N,N,N′,N′-tetra-p-phenylenediamine dihydrochloride (TMPD), phenol, hydrogen peroxide, DMSO, indomethacin, salicylhydroxamic acid, salicylic acid, sodium citrate, and thrombin receptor activating peptide were all obtained from Sigma Chemical (St. Louis, MO).
Site-Directed Mutagenesis.
Mutants of COX-1 and -2 were prepared using the QuikChange site-directed mutagenesis kit supplied by Stratagene (La Jolla, CA) as described previously (Loll et al., 2001). The R106Q mutant of COX-2 was prepared using the oligonucleotide primer 5′-G AGT TAT GTG TTG ACA TCC CAA TCA CAT TTG ATT GAC AGT CCA CC-3′.
Activity Assay.
Cyclooxygenase activity was measured using a coupled cyclooxygenase-peroxidase assay, monitoring the oxidation of TMPD at 611 nm after the addition of arachidonic acid (Tam et al., 1995). For inhibition studies, aliquots of stock solutions of compounds in DMSO were incubated with 1.1 μg of COX-1 for a specified time in 1 ml of 100 mM Tris-Cl, pH 8.0, and 1 μM hematin. TMPD (200 μM) was then added, followed by 40 μM arachidonic acid to start the reaction. Time-course experiments were carried out by prewarming enzyme solutions to 37°C, adding TriAcSHA at time 0, and then withdrawing aliquots for assays at various time points. Enzyme activities were normalized to the activity of enzyme solutions containing the vehicle alone.
Transient Expression of Human COX-1 and COX-2.
Wild-type and mutant pcDNA3-hCOX-1 constructs were transiently expressed in COS-1 cells. Parental vector pcDNA3 was used as a control for all transfection experiments. COS-1 cells were grown in 2 ml of Dulbecco's modified Eagle's medium, 10% fetal bovine serum, 100 units of penicillin, and 100 μg/ml streptomycin in six-well plates at 37°C, 5% CO2. Cells were transfected when 50 to 80% confluent. Purified plasmid DNA (1.5 μg) was transfected into COS-1 cells using 10 μl of LipofectAMINE (Invitrogen) for 24 h, as described in the manufacturer's protocol.
Assay of Prostaglandins Synthesized by Transfected COS-1 Cells.
Forty-eight hours after transfection the cells were assayed for COX-1 activity by enzyme immunoassay of PGE2(Assay Designs, Ann Arbor, MI).
Platelet Aggregation.
Platelet aggregation was measured turbidimetrically in platelet-rich plasma using a platelet aggregometer (model PAP-4; Biodata Corp., Horsham, PA). Aliquots of stock solutions of compounds in DMSO were incubated with 495 μl of platelet-rich plasma for 3 min before addition of the agonists arachidonic acid (1.5 mM) and TRAP (10 μM).
Synthesis of Inhibitors.
TriAcSHA was prepared as follows. Acetic anhydride (50 ml, 0.5 mol) and concentrated sulfuric acid (3 drops) were added to salicylhydroxamic acid (10.14 g, 66 mmol). The mixture was stirred at 80°C under nitrogen for 48 h, and ethyl acetate (100 ml) was added to an aliquot (10 ml) of the resulting dark-orange solution. The organic phase was washed twice with 0.5 M sodium hydroxide (50 ml), water (100 ml), 0.5 M citric acid (50 ml), and brine (100 ml). The organic layer was dried over MgSO4 and evaporated. The residue was purified by column chromatography on silica, using a dichloromethane/petroleum ether (3:2) eluant, giving a colorless oil (2.23 g, 8 mmol, 12%) and confirmed to be the desired compound by microanalysis and NMR spectroscopy. Microanalysis: C13H13NO6; required, C 55.92, H 4.69, N 5.02%; found, C 55.30, H 4.64, N 5.04%. 1H NMR (CDCl3); δ 1.9 (s, 3H, CH3), 2.18 (s, 3H, CH3), 2.46 (s, 3H, CH3), 7.16 (m, 2H, aromatic), 7.38 (m, 2H, aromatic). 13C NMR (CDCl3); δ 17.37 (s, 1C, CH3), 20.71(s, 1C, CH3), 24.71 (s, 1C, CH3), 123.03 (s, 1C, aromatic), 125.57 (s, 1C, aromatic), 126.29 (s, 1C, aromatic attached to subst. carbonyl), 128.72 (s, 1C, aromatic), 132.02 (s, 1C, aromatic), 147.10 (s, 1C, aromatic attached to subst. acetate), 165.62 (s, 1C, C=O), 166.47 (s, 1C, C=O), 167.14 (s, 1C, C=O), 168.52 (s, 1C, C=O).
The other tri-acyl derivatives of SHA were similarly prepared using the appropriate acid anhydride and also characterized by NMR spectroscopy (1H and 13C). The new compound N-Me-O,O-DiAcSHA was synthesized by treating acetylsalicylic acid (2 g; 11.1 mmol) withN-methyl hydroxylamine hydrochloride (0.92 g; 11.1 mmol) in the presence of N-methyl morpholine (2.34 ml; 33.3 mmol) and 2-chloro-4,6-dimethoxy-(1,3,5)triazine (2.34 g; 13.3 mmol) as described previously (De Luca et al., 2001). After work-up, this gaveN-methyl-O-acetylsalicylhydroxamic acid (an orange liquid), which was further acetylated with acetic anhydride as described above. 2-OMe-N,O-diacetylbenzohydroxamic acid was prepared according to a published method (Hosangadi and Dave, 1996). N,O-DiAcBHA was synthesized by acetylation of benzohydroxamic acid (73 mmol; 1 eq), in the presence of sulfuric acid. All compounds were characterized by microanalysis and NMR spectroscopy (1H and 13C).
Nomenclature.
The numbering of amino acids differs between ovine and human COX-1. Functional experiments employed both the human and ovine enzymes. To avoid confusion, we use the human numbering scheme throughout this article.
Results
Synthesis and Characterization of Acetylated Derivatives of SHA.
Treatment of SHA with acetic anhydride at 80°C for 48 h gave the desired compound, TriAcSHA, as a colorless oil. The product was characterized by microanalysis and by 1H and 13C NMR spectroscopy. The 1H spectrum in CDCl3 showed acetyl group signals at 1.9, 2.18, and 2.46 δ and aromatic signals at 7.16 and 7.38 ppm, all integrating for the required number of protons (Fig. 1). The13C NMR spectrum showed methyl group signals at 17.37, 20.71, and 24.71 ppm, six aromatic signals in the range 123.03 to 147.1, and four carbonyl group signals in the range 165.62 to 168.52 ppm. The purity of the diacetylated salicylhydroxamates was also confirmed by proton and 13C NMR, which showed the expected signals for these compounds. Additional triacyls of SHA were prepared similarly and characterized by 1H and13C NMR spectroscopy.
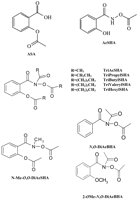
Structures of compounds.
TriAcSHA Inhibits COX-1 and COX-2.
COS-1 cells were transfected with human COX-1 or pcDNA3 alone and assayed for PGE2 production. Sham-transfected cells did not metabolize arachidonic acid, whereas cells expressing COX-1 synthesized high levels of PGE2 (1933 ± 276 ng/mg protein) (Fig. 2A). TriAcSHA (1 μM) reduced PGE2 to 147 ± 44 ng/mg of protein, whereas 1 μM ASA had little or no effect. At 10 μM, both TriAcSHA and ASA decreased PGE2 synthesis (to 29.7 ± 9.0 and 190 ± 60 ng/mg, respectively). As with aspirin, TriAcSHA displayed some selectivity for COX-1 (Fig. 2). Treatment with 10 μM TriAcSHA for 15 min caused 99% inhibition of COX-1 but only 76% inhibition of COX-2. The inhibition of COX-1 by TriAcSHA was both concentration- and time-dependent (Fig.3). Preincubation of COX-1 with inhibitor for 5 min yielded IC50 values of 18 μM for TriAcSHA, and 60 μM for ASA. Inhibition proceeded rapidly with the higher concentrations of TriAcSHA, with complete inhibition within 5 min at a concentration of 100 μM (Fig. 3).
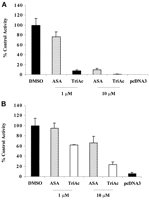
Inhibition of wild-type human COX-1 and COX-2 by TriAc and ASA. COS-1 cells were transfected with pcDNA3 constructs encoding either human COX-1 (A) or COX-2 (B) and with sham constructs of pcDNA3 alone. Cells were treated with 1 and 10 μM ASA and TriAcSHA, respectively, as well as the DMSO vehicle alone, for a period of 15 min. The cells were assayed for PGE2biosynthetic activity 15 min after the addition of arachidonic acid 60 μM. PGE2 levels were corrected for protein (nanograms per milligram of total protein) and expressed as a percentage of control (mean ± S.E.M., n = 3).
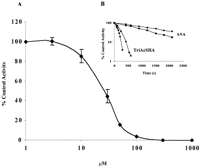
Inhibition of ovine COX-1 is dose- and time-dependent. A, purified enzyme was incubated with the inhibitor at 37°C for 5 min. The reaction was initiated by addition of 40 μM arachidonic acid, and activity of the enzyme was measured by monitoring the oxidation of TMPD. Enzyme activity is expressed as a percentage of the control. B, ovine COX-1 was incubated with inhibitor at 37°C; aliquots were withdrawn and cyclooxygenase activity measured at the times indicated (mean ± S.E.M., n = 3). ●, 50 μM aspirin; ▪, 100 μM aspirin; ▴, 50 μM TriAcSHA; ♦, 100 μM TriAcSHA.
Ser-529 Is Required for TriAcSHA Inhibition of COX-1 Activity.
COS-1 cells transfected with the S529A mutant of human COX-1 were incubated with 250 μM of either TriAcSHA or ASA for 45 min and PGE2 measured after the addition of 60 μM arachidonic acid. In contrast to cells transfected with the wild-type enzyme, neither TriAcSHA (331 ± 12 ng/mg protein) nor ASA (247 ± 45 ng/mg protein) inhibited product formation by the S529A mutant (266 ± 27 ng/mg protein).
Active Site Arginine in the Inhibition of COX-1 and COX-2 by TriAcSHA.
The positively charged arginine residue (R119 in COX-1, R106 in COX-2) located at the neck of the cyclooxygenase active site is required for the binding of arachidonate and the activity of aspirin and several other NSAIDs. To investigate whether this arginine was required for the activity of TriAcSHA, COS-1 cells were transfected with the R119Q mutant of COX-1 or the R106Q mutant of COX-2. The R119Q COX-1 but not the R106Q COX-2 produced much lower levels of PGE2 than the wild-type enzyme (Fig.4). Mutating the R119 in COX-1 markedly reduced the potency of TriAcSHA. Thus, incubating the enzyme with 10 μM TriAcSHA for 15 min decreased PGE2 synthesis by only 40% (10.1 ± 5.0 to 6 ± 2.4 ng/mg of protein). Increasing the concentration of TriAcSHA to 250 μM for 45 min abolished PGE2 synthesis. Mutating the R106 of COX-2 also attenuated the effect of TriAcSHA compared with the wild-type enzyme (Fig. 4).
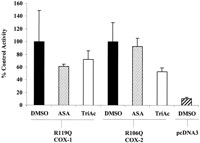
Role of the active site arginine in the inhibition of COX-1 and COX-2 by TriAcSHA. COS-1 cells were transfected with the R119Q mutant of COX-1 or the R106Q mutant of COX-2. Cells were treated with 10 μM ASA or TriAcSHA as well as the DMSO vehicle alone for a period of 15 min. The cells were assayed for PGE2 15 min after the addition of 60 μM arachidonic acid (mean ± S.E.M.,n = 3).
TriAcSHA Inhibits Platelet Aggregation.
TriAcSHA inhibited arachidonic acid-induced platelet aggregation with an IC50 value of 10.2 ± 3 μM compared with 53.3 ± 3.2 μM for aspirin (Fig.5). Both compounds exhibited time-dependent inhibition. ASA (30 μM) completely inhibited arachidonic acid-mediated platelet aggregation by 15 min, whereas no inhibition was evident at 5 min. At 30 μM, TriAcSHA completely inhibited platelet aggregation within 5 min. As expected, TriAcSHA did not inhibit platelet aggregation induced by 10 μM thrombin receptor activating peptide, where the response is independent of COX activity.
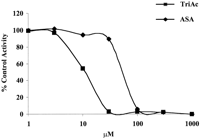
TriAcSHA inhibits platelet aggregation. Increasing concentrations of ASA and TriAcSHA (TriAc) were incubated with human platelet-rich plasma for 5 min before the addition of 1.5 mM arachidonic acid. Platelet aggregation was measured as a percentage of the response in platelets treated with vehicle (DMSO) alone (mean ± S.E.M., n = 3).
Diacetylated Derivatives of AcSHA.
To explore the mechanism of acetylation, we synthesizedN-Me-O,O-DiAcSHA,N,O-DiAcBHA, and 2-OMe-N,O-DiAcBHA.N-Me-O,O-DiAcSHA showed moderate inhibitory activity (IC50 170 μM) in platelet rich plasma (COX-1), whereas N,O-DiAcBHA, and 2-OMe-N,O-DiAcBHA were inactive, suggesting that the enzyme is covalently modified by the phenolic acetyl.
Triacyl Analogs of TriAcSHA Are Selective for COX-1.
TriAcSHA was used as a template for the synthesis of a second generation of COX inhibitors where the three acyl side chains were sequentially lengthened by one carbon to give the tripropyl, tributyl, trivaleryl, and trihexyl analogs (Fig. 1). All compounds inhibited COX-1 expressed in COS-1 cells, with increasing selectivity for COX-1 as the side chain lengthened (Fig. 6). Preincubation of COX-1 with inhibitors for 30 min gave IC50 values that decreased with increasing side chains: 580 ± 54 μM for tripropyl, 20.2 ± 2.7 μM for trivaleryl and 4.75 ± 0.7 μM for trihexyl (mean ± S.E.M., n = 3). There was no inhibition of COX-2 expression in COS-1 cells with 250 μM trihexyl.
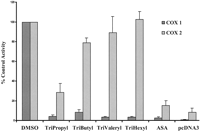
Inhibition of COX-1 and COX-2 by triacyl analogs of TriAcSHA. COS-1 cells transfected with human COX-1 or COX-2 were treated with 250 μM of the triacyl analog for 15 min and the supernatant was assayed for PGE2 15 min after the addition of 60 μM arachidonic acid (mean ± S.E.M., n= 3).
Discussion
Aspirin is unique among NSAIDs in reducing the risk of thrombosis in patients with cardiovascular disease. This may be explained by its mechanism of action. Only aspirin irreversibly inactivates the enzyme cyclooxygenase. Because the anucleate platelet cannot synthesize new enzyme, complete suppression of platelet thromboxane formation is achieved with a single daily dose of aspirin. Moreover, because platelets have a lifespan in the circulation of 10 days, a small daily dose of aspirin is sufficient to maintain complete suppression of platelet cyclooxygenase. Other NSAIDs are reversible inhibitors of cyclooxygenase, so that platelet activity recovers between doses.
Despite these advantages, few acylating drugs such as aspirin have been generated. Those that have deliver the acyl group from the same position relative to the carboxylic acid, limiting the potential for generating novel compounds. We have synthesized a compound, AcSHA (O'Brien et al., 1997), in which the acetyl is attached to a hydroxamate group. This compound inactivated COX-1 in the same fashion as aspirin by acetylating the serine 529 within the cyclooxygenase pocket (Loll et al., 2001). Although the compound was less potent than aspirin, AcSHA provided a backbone for the generation of other acylating compounds. In this study, we showed that one of these, TriAcSHA, which is structurally very different from aspirin, was more effective than aspirin in inactivating COX and inhibiting platelets.
Like aspirin, the activity of TriAcSHA was dependent on the presence of the serine residue 529, mutation of which ablated the drug's activity. The activity of TriAcSHA was also dependent on arginine 119; mutation of this residue markedly attenuated its inhibitory effect. In the case of aspirin, the positively charged guanidinium side chain of this arginine residue interacts with the carboxylate group of aspirin. Indeed, the crystal structure of COX-1 treated with bromoaspirin shows that the salicylate metabolite forms a salt-linkage with this residue (Loll et al., 1995). Similarly, the crystal structure of the AcSHA/COX-1 complex shows that the inhibitor forms a salt-linkage with the arginine 119 side chain through the deprotonated nitrogen of the acetylated hydroxamate group (Loll et al., 2001).
Of the three acetyl groups in TriAcSHA, the O-hydroxamate acetyl group can be ruled out as the acetylating moiety, because high-performance liquid chromatography analysis, after incubation of COS-1 cells with TriAcSHA for 45 min, found only AcSHA, which is much less active than the TriAcSHA. This suggests that either theN-(hydroxamate) acetyl group or the phenolic acetyl group are responsible for the acetylation of the serine residue. To establish the source of acetylation, we synthesized a series of structurally related compounds to investigate their inhibitory effects. Based on their lack of activity (see below), we propose that the interaction between R119 and TriAcSHA, which (unlike ASA and AcSHA) does not contain an acidic group, is caused by a hydrogen bonding interaction between its guanidinium side chain and the oxygen of theN-acetyl group. This interaction may then cause general acid-catalyzed N-deacetylation, givingO,O-diacetylsalicylhydroxamate (O,O-DiAcSHA) in the active site pocket. This species, which is likely to form a salt-linkage with R119 through its deprotonated nitrogen [the pKa value for this compound should be lower than 5.6 (i.e., that reported for AcSha)], is highly labile; the negatively charged nitrogen provides intramolecular assistance to acetyl group transfer from the phenolic group to Ser 529. A similar labilization of a phenolic acetyl group by a deprotonated nitrogen of an amide was reported in the decomposition of O-acyl derivatives of salicylamide (Tawfiq et al., 1990). Evidence for the high lability of the acetylated phenolic group in the proposed O,O-DiAcSHA intermediate is provided by the fact that it could not be isolated; in aqueous solution, TriAcSHA hydrolyzes to AcSHA without observation of any diacetylated species. (Under physiological conditions, hydrolysis of TriAcSHA to AcSHA is slow; after 10 min, 100% of TriAcSHA is present, reducing to 87% TriAcSHA and 13% AcSHA after 40 min.)
Consistent with this proposed mechanism,N-methyl-O,O-DiAcSHA, which does not contain an ionizable -NH group, shows greatly reduced activity. Further evidence that the phenolic acetyl group is the acetylating moiety is provided by the lack of activity of the structurally related diacetylated compounds N,O-DiAcBHA and and 2-OMe-N,O-DiAcBHA both of which contain hydroxamate N-acetyl groups but lack acetylated phenolic groups.
We used the same synthetic strategy as that used for TriAcSHA to generate a series of triacyl compounds with progressively longer acyl groups, all of which inhibited COX. In previous work, Bhattacharyya et al. (1995) showed that among a series of monoacylated salicylates, only valerylsalicylic acid was selective for COX-1. The salicylhydroxamate scaffold allows a greater range of acyl groups to be delivered. As with valerylsalicylic acid, increasing the number of carbons resulted in greater selectivity for COX-1 over COX-2, a surprising finding because the channel in COX-2 is larger (Luong et al., 1996). However, unlike the monoacylated salicylates, these compounds show increased potency with increasing length of the acyl group, with the trihexyl compound exhibiting an IC50 of 5 μM and a surprisingly high degree of selectivity for COX-1.
Selective inactivation of COX-1 may provide advantages over aspirin as antithrombotic therapy. Aspirin is effective as a platelet inhibitor when given in a low daily dose. However, coincident inhibition of COX-2 may limit its effectiveness. Recent studies show that COX-2 is responsible for up to 80% of the prostacyclin generated in man (Belton et al., 2000). Prostacyclin, a platelet inhibitor, is largely derived from vascular endothelium and in vivo attenuates the response to thromboxane, which is largely derived from COX-1 in platelets (Cheng et al., 2002). Thus, through inhibition of COX-2, the antiplatelet effect of aspirin may be offset by coincident inhibition of prostacyclin. Inhibition of COX-2 in the gut may also play a role in the gastrointestinal injury seen with even low doses of aspirin (Hudson et al., 1992). Although the damage to gastric mucosa with aspirin and other NSAIDs was originally attributed to COX-1 inhibition, experimental evidence implicates both isoforms (Wallace et al., 2000). For example, in the COX-1 knockout mouse, gastric injury is absent in the untreated animal but is induced by nonselective NSAIDs and by selective COX-2 inhibitors (Langenbach et al., 1995). Indeed, selective COX-1 inhibitors do not induce gastric injury in experimental models.
In conclusion, we have shown that salicylhydroxamic acid provides a versatile backbone for the generation of a family of acylating inhibitors of cyclooxygenase.
Footnotes
-
Financial support was provided by the Health Research Board, The Research Committee of the Royal College of Surgeons, Enterprise Ireland, and Higher Education Authority of Ireland.
- Abbreviations:
- COX
- cyclooxygenase
- NSAID
- nonsteroidal anti-inflammatory drug
- ASA
- acetylsalicylic acid (aspirin)
- SHA
- salicylhydroxamic acid
- AcSHA
- acetylsalicylhydroxamic acid
- TriAcSHA
- triacetylsalicylhydroxamic acid
- TMPD
- N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride
- DMSO
- dimethyl sulfoxide
- PG
- prostaglandin
- O,O-DiAcSHA
- O,O-diacetylsalicylhydroxamic acid
- N,O-DiAcBHA
- N,O-diacetylbenzohydroxamic acid
- N,O-DiAcSHA
- diacetylsalicylhydroxamic acid
- 2-OMe-N,O-DiAcBHA
- 2-methoxy-N,O-diacetylbenzohydroxamic acid
-
- Received July 17, 2002.
- Accepted November 6, 2002.
- The American Society for Pharmacology and Experimental Therapeutics



