

Structural Basis of Enantioselective Inhibition of Cyclooxygenase-1 by S-α-Substituted Indomethacin Ethanolamides*
Back to the Index...COX- Christine A. Harman‡,1,
- Melissa V. Turman§,1,2,
- Kevin R. Kozak§,
- Lawrence J. Marnett§,
- William L. Smith¶ and
- R. Michael Garavito‡,3
+ Author Affiliations
- ↵3 To whom correspondence should be addressed: Dept. of Biochemistry and Molecular Biology, Michigan State University, 513 Biochemistry, East Lansing, MI 48824. Tel.: 517-355-9724; Fax: 517-353-9334; E-mail: garavito@msu.edu.
Abstract
The modification of the nonselective nonsteroidal anti-inflammatory drug, indomethacin, by amidation presents a promising strategy for designing novel cyclooxygenase (COX)-2-selective inhibitors. A series of α-substituted indomethacin ethanolamides, which exist as R/S-enantiomeric pairs, provides a means to study the impact of stereochemistry on COX inhibition. Comparative studies revealed that the R- and S-enantiomers of the α-substituted analogs inhibit COX-2 with almost equal efficacy, whereas COX-1 is selectively inhibited by the S-enantiomers. Mutagenesis studies have not been able to identify residues that manifest the enantioselectivity in COX-1. In an effort to understand the structural impact of chirality on COX-1 selectivity, the crystal structures of ovine COX-1 in complexes with an enantiomeric pair of these indomethacin ethanolamides were determined at resolutions between 2.75 and 2.85Å. These structures reveal unique, enantiomer-selective interactions within the COX-1 side pocket region that stabilize drug binding and account for the chiral selectivity observed with the (S)-α-substituted indomethacin ethanolamides. Kinetic analysis of binding demonstrates that both inhibitors bind quickly utilizing a two-step mechanism. However, the second binding step is readily reversible for the R-enantiomer, whereas for the S-enantiomer, it is not. These studies establish for the first time the structural and kinetic basis of high affinity binding of a neutral inhibitor to COX-1 and demonstrate that the side pocket of COX-1, previously thought to be sterically inaccessible, can serve as a binding pocket for inhibitor association.
Cyclooxygenase (COX4; also known as prostaglandin endoperoxide synthase) is a bifunctional enzyme that catalyzes the conversion of arachidonic acid to prostaglandin (PG) H2, the immediate precursor to prostaglandins, thromboxane, and prostacyclin. The conversion of arachidonic acid into PGH2 proceeds through two separate reactions in which two molecules of O2 are incorporated into arachidonic acid bound in the COX site to form PGG2, which then diffuses to the peroxidase site (POX) to undergo a two-electron reduction to form the final product PGH2. Two isoforms of COX exist and are referred to as COX-1 and COX-2. COX-1 is constitutively expressed in many tissues to produce the amounts of prostaglandins needed for regulating normal “housekeeping” functions such as renal water retention, gastric acid secretion, parturition, and hemostasis. In contrast, various inflammatory stimuli associated with the immune response rapidly induce the expression of COX-2 and the associated prostaglandins, which result in pain, fever, and inflammation (1).
Nonsteroid anti-inflammatory drugs (NSAIDs) provide therapeutic benefit through the direct inhibition of the COX enzymes; however, the anti-inflammatory and analgesic properties of NSAIDs are mainly a result of inhibition of COX-2. Although nonselective inhibition of COX-1 by aspirin can be beneficial in reducing the risk of mortality from cardiovascular disease, the use of aspirin and other NSAIDs has been associated with undesirable gastrointestinal side effects such as stomach ulcers and intestinal bleeding (2, 3). The predominant role of COX-2 in pain and inflammation spawned the development of selective inhibitors, which target only the COX-2 isoform to achieve analgesic and anti-inflammatory properties with minimal associated gastrointestinal toxicity. However, cardiovascular side effects have recently been documented for at least certain COX-2 inhibitors (e.g. rofecoxib, valdecoxib, and celecoxib).
Structural and functional studies comparing COX-1 and COX-2 have identified some general structural features of NSAIDs binding to the COX active site, in addition to highlighting subtle biochemical differences between the isoforms that have been exploited in the development of COX-2-selective inhibitors (4-6). The entrance to the COX active site is made up in part by a bundle of four amphipathic helices, which leads to a constriction comprised of residues Arg-120,5, Tyr-355, and Glu-524. The COX active site then opens into a long hydrophobic channel that extends deep into the core of the catalytic domain of the protein. Sitting above the constriction and aside the main hydrophobic channel is a smaller amphipathic region referred to as the “side pocket.” A major difference in the active site of the isoforms arises mainly as a result of a valine substitution in COX-2 for isoleucine in COX-1 at position 523. This small change provides additional space to allow better access to the side pocket of COX-2. The active site of COX-2 can therefore accommodate the extra bulk of the sulfonamoylphenyl or methylsulfonylphenyl moieties of COX-2-selective inhibitors, particularly those of the diarylheterocycles (celecoxib and rofecoxib) by placing them in the side pocket.
The potential for interaction between moieties of the drug and residues within the side pocket of COX-2 also seems to be partly responsible for time-dependent inhibition by COX-2-selective NSAIDs. Although diarylheterocycles competitively inhibit COX-2 better than COX-1, their lack of time-dependent inhibition behavior toward COX-1 further enhances their selectivity toward COX-2 (7, 8). The structural explanation for the lack of time-dependent inhibition toward COX-1 is the extra methylene group of isoleucine at position 523, which restricts access of COX-2 inhibitors to the side pocket in COX-1.
COX-2-selective inhibitors also lack a free carboxylate group, which may contribute to their low affinity toward COX-1. Several crystal structures of COX-1 and COX-2 have shown that NSAIDs with a free carboxylate moiety form an ionic interaction with Arg-120 at the mouth of the COX active site (4, 6, 9, 10). This interaction between the carboxylate of acidic NSAIDs and Arg-120 appears to be essential for substrate binding to COX-1, as site-directed mutagenesis studies of COX-1 have demonstrated that the binding of arachidonic acid and acidic NSAIDs was greatly perturbed when Arg-120 is mutated to smaller or uncharged residues (11). In contrast, the equivalent mutations of Arg-120 seemed to suggest that electrostatic interactions with this residue in COX-2 are less critical for catalysis and NSAID inhibition (12, 13).
Modification of the carboxylate group of the nonselective NSAID indomethacin generates a wide range of ester and amide adducts yielding an array of COX-2-selective inhibitors (14). The majority of these indomethacin derivatives was shown to be potent and selective for COX-2; however, an exception was observed with the α-substituted indomethacin ethanolamide series of derivatives (15). The (R/S)-α-substituted indomethacin ethanolamides both demonstrated potent inhibition of COX-2; however, the S-α-substituted indomethacin ethanolamides inhibited COX-1 as effectively as COX-2. Interestingly, the chiral preference observed with COX-1 inhibition was consistently observed with S-enantiomers across a wide range of α-substitutions (Table 1). Mutagenic studies in COX-2 were unable to identify residues around the COX active site that contributed to the observed chiral selectivity. To understand the structural basis of this chiral discrimination in COX-1, we determined the crystal structures of ovine COX-1 complexed with the α-ethyl-substituted enantiomeric pair of these indomethacin ethanolamides. In addition, the kinetic basis of selectivity was examined using pre-steady state measurements. These structures not only provide intriguing insight into the structural basis for the stereoselective binding of the S-α-substituted indomethacin ethanolamides to COX-1, but, in conjunction with kinetic analysis provide an illustration of how COX-1 can bind a noncarboxylate containing inhibitor with high affinity.
Comparative inhibition of native COX-1 and COX-2 by various α-substituted indomethacin ethanolamides
Data shown in table were obtained from a previously published study by Kozak et al. (15).
EXPERIMENTAL PROCEDURES
Materials—Detergents C10E6 used for solubilization and n-octyl-β-d-glucopyranoside (β-OG) used for purification and crystallization were purchased from Anatrace (Maumee, OH). Hemin was purchased from Sigma. Compound 8 (indomethacin-(R)-α-ethyl-ethanolamide) and compound 9 (indomethacin-(S)-α-ethyl-ethanolamide) used in crystallization experiments were synthesized according to the procedures described in Kozak et al. (15). The nomenclature as 8 and 9 refers to their earlier designation (15).
Crystallization of Native Ovine COX-1-Inhibitor Complexes—COX-1 used for crystallization was purified from ram seminal vesicles as described previously (16). Protein was prepared for crystallization trials by adding 1.5 m excess of hemin to reconstitute the protein. Inhibitors at concentration of 100 μm of either 86 or 9 were then added to protein and left to incubate for at least 10 min before setting up crystallization experiments. Using the sitting drop vapor diffusion method, 3 μl of protein was mixed with 3 μl of buffer composed of 0.64 m sodium citrate, 0.3-0.9 m LiCl, 0.3% (w/v) β-OG, and 1 mm NaN3 and was equilibrated within a reservoir containing 0.68-0.88 m sodium citrate, 0.3-0.6 m LiCl, and 1 mm NaN3. Crystals appeared within 2-3 weeks.
Data Collection of oCOX-1·8 and oCOX-1·9 Complexes—Crystals were harvested, briefly soaked in a solution containing 1.0 m sodium citrate, 1.0 m LiCl, and 0.15% (w/v) β-OG, and 1 mm sodium malonate as a cryoprotectant. Crystals were then flash-frozen in liquid nitrogen at -165 °C. Data collection on crystals of COX-1-drug complexes was performed at beamline 5-ID at COM-cat (Argonne National Laboratory, Argonne, IL). Data from a single crystal of COX-1 complexed with 8 (COX-1·8) was indexed and integrated using DENZO (17) and then scaled with SCALEPACK (17). Data sets collected from two crystals of COX-1 complexed with 9 (COX-1·9) were indexed and integrated separately and then merged together during scaling using SCALEPACK (Table 2).
Summary of data collection and refinement statistics
Structure Determination and Refinement of oCOX-1·8 Complex—The COX-1 structure complexed with 8 was determined using native COX-1 structure (Protein Data Bank entry 1DIY) as a phasing model for rigid body refinement utilizing CNS version 1.1 (18). After an initial round of simulated annealing, several sugar moieties, detergent molecules, and the inhibitors were fitted to density in several iterative cycles of positional refinement, group B-factor refinement, and manual model building. To further complete the model, water molecules were added at positions in the Fo - Fc difference electron density maps where the peak heights were greater than 2.5σ and within 2.4 -3.6 Å of a hydrogen bond partner. At final round of refinement, the R and Rfree converged 24.5 and 29.2%.
Structure Determination and Refinement of COX-1·9 Complex—The COX-1·9 was refined with REFMAC version 5.0 in the CCP4 program suite (19). REFMAC refinement started with the protein portion of using native COX-1 structure (Protein Data Bank entry 1DIY). The COX-1·9 structure was initially put through TLS refinement (20-21) and then restrained positional refinement. Each round of refinement included 10 cycles of TLS, followed by 20 cycles of restrained refinement (which included positional refinement followed by B-factor refinement). A single TLS group consisting of protein residues 33-580 was used for the TLS portion of refinement. Iterative cycles of TLS/restrained refinement were followed by cycles of map calculation and model building. In successive rounds of refinement, the heme group, six saccharide moieties, a glucopyranoside head group from β-OG, and ∼20 water molecules (using the same criteria used for oCOX-1·8 complex) were added to the model. At the final round of refinement, the R and Rfree converged at 24.1 and 29.2%.
Modeling of Compound 8 Using Simulated Annealing—Five rounds of simulated annealing utilizing molecular dynamics in CNS version 1.1 (18) were performed using an initial annealing temperature of 2500 K, the observed structure factors of the COX-1·9 (protein only), and a starting model of COX-1·9 protein and compound 8 with the ethanolamide moiety oriented in side pocket of the COX-1 active site.
Time Dependence of oCOX-1—Inhibition Apo-COX-1 (50 nm) was reconstituted with 2.5 eq of hemin in 100 mm Tris-HCl (pH 8.0) containing 500 μm phenol. Reconstituted enzyme was preincubated at 37 °C with either 100 μm 8 or 1.75 μm 9 for various lengths of time. Following preincubation, 50 μm [14C]arachidonic acid was added, and the incubation was allowed to proceed for 30 s at 37 °C. Reactions were terminated and analyzed for substrate consumption by thin layer chromatography as described previously (14).
Reversibility of oCOX-1 Inhibition—COX-1 was reconstituted as described above and was preincubated at 37 °C with Me2SO (2.5% final v/v) or 50 μm 8 or 9 for 3 min at 37 °C. Cyclooxygenase reactions were initiated by the addition of 50 μm [14C]arachidonic acid. Aliquots were removed at specific time points (30 s to 5 min) and transferred to tubes containing termination solution. Substrate consumption was analyzed by thin layer chromatography as described previously (14).
Pre-steady State Kinetics of Inhibitor Binding to oCOX-1—Pre-steady state kinetic measurements were carried out in 25 mm sodium phosphate (pH 7.4) using 200 nm apo-COX-1 and varying inhibitor concentrations (1-25 μm). Measurements were acquired on an Applied Photophysics Ltd. model SX17MV (United Kingdom) stopped-flow fluorometer with a 1-cm path length cell. Quenching of intrinsic protein fluorescence was monitored following excitation at 280 nm. An average of four to eight shots was taken for each inhibitor concentration, and vehicle control was subtracted to correct for nonspecific photobleaching. Resulting traces were fit to single exponential decay using ProK software (Applied Photophysics Ltd.) to extract amplitudes and observed rate constants (kobs). For each inhibitor, kobs was plotted against concentration of inhibitor, and the data fit by nonlinear regression.
RESULTS AND DISCUSSION
Binding of 8 Versus 9 in the COX Site—Compounds 8 and 9 were modeled into the electron density maps with the ethanolamide moiety in the lower energy trans-amide bond configuration. The observed electron density for both 8 and 9 was not clear enough at 2.8 Å resolution to determine whether the amide bond adopted either a cis or trans configuration with a high degree of certainty. The trans conformer for the ethanolamide moiety is most likely to be the stable conformer in solution (32). Moreover, the transition between the trans and cis conformers has to overcome a high activation energy barrier. Nonetheless, the cis-amide conformer could not be unequivocally ruled out. Thus, for completeness, the cis-amide bond configuration for both 8 and 9 was also examined to determine whether it impacted the interactions of the ligands within the COX active site.
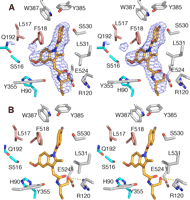
Compound 8 bound in the COX active site of COX-1. A, stereo view of 8 bound in COX active site with simulated annealing omit map difference density (blue) contoured at 3σ witha3Å boundary around the ligand. Various residues within the active site are shown with carbons for 8 shown in orange, oxygen red, nitrogen blue, and chlorine purple. B, stereo representation of 8 bound in the COX site in the same manner as the parent compound indomethacin; the chlorobenzoyl group is oriented up toward the top of the channel, the methoxy group on indole ring points toward the side pocket (Leu-517, Phe-518, Ile-523, Gln-192, and Ser-516), and the ethanolamide group with the R-ethyl substitution sits next to Arg-120 and Tyr-355 at the mouth of the active site. Carbon atoms of 8 are shown with same color scheme as in A with the yellow dashed lines representing various interactions (hydrophilic) between ligand and protein residues. The hydroxyl group of the ethanolamide group makes a hydrogen bond with Arg-120 and Glu-524, whereas the (R)-ethyl group is positioned just outside the mouth of the active site. Refinement statistics for both structures are shown in Table 2. All figures presented here were created using the program PyMOL.
The electron density maps obtained for the COX-1·8 structure shows that the R-stereoisomer binds in an orientation that closely resembles the binding mode of the parent compound indomethacin (4, 9); the chlorobenzoyl group is oriented up to the apex of the channel by Tyr-385, the methoxy group of the indole ring points toward the side pocket, and the ethanolamide group is positioned toward the mouth of the channel near Arg-120 and Tyr-355 (Fig. 1, A and B). The hydroxyl group of the ethanolamide moiety of 8 makes a hydrogen bond with the guanidinium group of Arg-120 and with carboxylate of Glu-524 (Fig. 1B). The trans conformer of 8 fits the electron density equally well before and after refinement. The final refined model generated no positive or negative density peaks observed around the inhibitor in the Fo - Fc difference electron density maps.
The interactions observed between residues of the COX site and ethanolamide moiety in the cis conformer were also essentially identical to the interactions generated with the trans conformer. However, some minor, but interesting differences were observed. With the amide bond in the trans configuration, the hydroxyl of the ethanolamide made an additional interaction with the carboxylate of Glu-524. In addition, the position of Arg-120 differed noticeably after refinement with the trans conformer of 8. Given no evidence to the contrary, 8 most likely binds to the COX-1 active site as the trans conformer, a conclusion that is consistent with the modeling study by Moth et al. (32).
After refinement of the COX-1·9 structure, the observed electron density for 9, the S-stereoisomer, revealed an elongated stretch of electron density that extends into the side pocket region of the COX active site (a region defined by residues Gln-192, His-90, Leu-517, Phe-518, and Ile-523 (Fig. 2A). The extra electron density within the side pocket is not observed in the structure of the COX-1·8 complex and suggests a different and contrasting mode of binding for the S-stereoisomer. After extensive model building trials, it is clear that 9 binds within the COX-1 active site with the chlorobenzoyl moiety oriented toward the mouth, the methoxy group pointed toward the top of the channel by Tyr-385, and the ethanolamide group positioned in the side pocket region (Fig. 2, A and B).
This conclusion is supported by an inspection of the Fo - Fc difference electron density after the final rounds of refinement. With the final refined model, no significant positive or negative electron density peaks were observed around compound 9, including around the region of the ethanolamide group. Some poorly defined negative difference density was observed near the chlorobenzoyl moiety. However, this peak was not well defined, was not centered on any particular atom, and was not particularly intense (<3σ). This feature is most likely an artifact arising from the medium resolution of data or some disorder of the inhibitor binding mode.
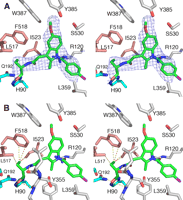
Compound 9 bound in the COX active site of COX-1. A, stereo view of 9 fitted into simulated annealing omit map difference density (blue) contoured at 3σ witha3Å boundary around the ligand. Carbon atoms of 9 are colored green with heteroatoms colored the same as in 1. B, chlorobenzoyl group of 9 is oriented toward the mouth of the active site; the methoxy group of indole ring is oriented up into the top of the channel, and the ethanolamide group with the (S)-ethyl substitution lies within the side pocket region of the channel. Yellow dashed lines represent various interactions (hydrophobic and hydrophilic) between ligand and protein. The hydroxyl group of the ethanolamide is oriented near the hydrophilic region (cyan) of the side pocket making hydrogen bonds with residues Gln-192 and His-90 whereas the S-ethyl group is oriented into the more hydrophobic region (pink) of the side pocket making van der Waals interactions with Ile-523, Phe-518, and Ile-517. For clarity, an additional polar interaction between Arg-120 and the carbonyl of the chlorobenzoyl moiety is not shown.
In this altered mode of binding, the ethanolamide group of 9 makes many interactions with hydrophilic and hydrophobic residues in the side pocket with the hydroxyl group making hydrogen bonds with His-90 and Gln-192. There are also several hydrophobic interactions involving Phe-518 and Ile-523 (Fig. 2B). An additional hydrophilic interaction is observed between the guanidinium group of Arg-120 and the carbonyl group of the chlorobenzoyl moiety positioned at the mouth of the active site. The trans configuration of the amide bond fits the electron density well, before and after refinement. Although the interactions observed between the ethanolamide moiety and the side pocket of COX site were maintained with either the cis or trans configuration, the trans configuration allows the formation of an additional interaction between the amide nitrogen of 9 and the carbonyl oxygen from the backbone of residue Leu-352. The observation supports the hypothesis that the trans conformer is the more prevalent conformer (Fig. 2B), when 9 binds to COX-1.
Evaluation of the Structural Basis for Stereoselectivity—The inhibitor complexes suggest that the distinctly different binding modes for 8 and 9 are the result of the S-stereoisomer making more favorable binding interactions within the side pocket compared with the R-stereoisomer, which has few interactions with the side pocket. The ethanolamide moiety of 9 utilizes both the hydrophilic and hydrophobic nature of groups within the COX-1 side pocket (i.e. residues Gln-192, His-90, IIe-523, Phe-518, and Leu-517) (Fig. 2B).
The binding mode of 9 is analogous to the binding of COX-2-selective inhibitors to COX-2, which suggests that the side pocket in COX-1 is accessible depending on the nature of the substituents present in the inhibitor. Structure-activity studies, which systematically evaluated various substitutions at the α-position indomethacin ethanolamides, found that altering the hydroxyl group influenced the chiral dependent selectivity toward COX-1 to some degree (15). When the hydroxyl group was replaced with either a methoxy or methyl group, the chiral dependence changed so that the R-enantiomers were more potent against COX-1 than the corresponding S-enantiomers. Collectively, these results suggest that the inability of the (R) enantiomers to inhibit COX-1 was not merely because of an increase in unfavorable steric interactions but also because of the loss of favorable binding interactions. The elaborate network of interactions needed to favor the more stable binding mode of 9 in the COX-1·9 crystal structure is consistent with this hypothesis. In contrast, the binding mode of 8 observed in the COX-1·8 crystal structure displays a quite different set of interactions, but none occur within the side pocket. Instead, the ethanolamide moiety of 8 is oriented toward the mouth of the active site, whereupon the ethanolamide hydroxyl makes hydrophilic interactions with the guanidinium group of Arg-120 and a hydroxyl group of Glu-524 (Fig. 1B). This mode of binding for 8 is virtually identical to that derived by Moth et al. (32) from a modeling study with indomethacin ethanolamide compounds having an (S)- and (R)-propyl substitution.
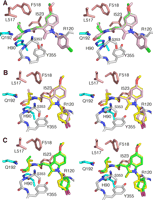
Comparison of compound 8 conformers obtained from simulated annealing and the placement of the (R)-ethyl versus the (S)-ethyl group in COX-1 side pocket. A, stereo view of all five conformers of 8 obtained from simulated annealing experiments are shown bound in the COX-1 active site. Four of the five conformers being virtually identical are shown with light purple carbons, whereas the conformer most similar to the binding configuration of 9 is shown with green carbons. With all five conformers, the hydroxyl group of the ethanolamide moiety makes ionic interactions with His-90 and Gln-192. Note how the orientation of the (R)-ethyl group for the four light purple conformers differs with respect to that of the green conformer. Color scheme of heteroatoms are nitrogen blue, oxygen red, and chlorine green. B, stereo view of 9 (shown with yellow carbons) and a representative of the four identical conformers of 8 (shown in light purple as in A) superimposed for comparison. Color scheme for heteroatoms are same as in A, except chlorine is purple. C, stereo view of 9 and a conformer of 8 being most like 9 (shown in green as in A) superimposed for comparison. Carbons of the 8 conformer are colored green, whereas carbons of 9 are colored yellow. Color scheme for heteroatoms is same as in B.
To visualize why compound 8 may not adopt the same binding mode as compound 9, compound 8 was modeled into the COX active site with the ethanolamide moiety positioned in the side pocket (Fig. 3A). The starting conformation of 8 also has the hydroxyl group of the ethanolamide positioned making favorable hydrophilic interactions with the Gln-192 and His-90. A representative set of conformers was then generated by performing five rounds of simulated annealing molecular dynamics using the protein shell of COX-1·9 complex and 8 as the model and the observed structure factors of the protein shell of COX-1·9 complex alone.
Four of the five conformers generated from simulated annealing had essentially the identical conformation (Fig. 3A). All five conformers had the hydroxyl group of the ethanolamide positioned to make favorable hydrophilic interactions with Gln-192 and His-90; however, the (R)-ethyl group of four of the five could not make the hydrophobic interactions observed in the 9 structure (Fig. 3B). Furthermore, with the (R)-ethyl and hydroxyl groups of 8 oriented toward the same side of the COX side pocket, little free space is available, and the potential for steric clashes increases. In contrast, one of the five conformers possessed a conformation, which orients the ethyl group and hydroxyl group on opposites sides of the side pocket, as observed for 9 (Fig. 3C).
Results of these simulated annealing studies demonstrate that there is no obvious structural explanation for why 8 cannot adopt the same binding mode of 9. The modeling revealed a conformer that can make similar interactions within the side pocket as seen with 9. However, these studies do suggest several insights as to why 8 does not adopt the binding mode of 9. Only one of the five conformers obtained from simulated annealing was able to achieve a similar conformation to 9. This suggests that 8, when bound like 9, does not have as favorable fit in the side pocket as 9. In contrast, the same simulated annealing experiments conducted on 9 generated only conformers identical to the refined structure of 9.
Another major difference between 9 and the conformers of 8 observed in the simulated annealing experiments is the orientation of the ethyl group with respect to the “roof” of the COX side pocket (Fig. 3). All five conformers of 8 have the (R)-ethyl group oriented up toward the roof. In contrast, the (S)-ethyl group of 9 is always oriented down toward the “floor” of the side pocket where there is more space to accommodate the entry of the ethyl group. Thus, the (R)-ethyl group of 8 may be able to fit within the side pocket, but the ability of 8 to access the side pocket may be prevented by lack of clearance. In particular, Phe-518 may play a role in obstructing the clearance of the (R)-ethyl group of 8; the (R)-ethyl group of the conformer most similar to 9 makes a very close van der Waals contact (<3.0 Å) with Phe-518 as compared with that observed in 9 (Fig. 3C). However, this explanation may suggest that the dynamics of inhibitor binding may be a more important factor in determining inhibitor stereoselectivity than the final binding conformation. As noted by Walker et al. (8), the kinetic studies of COX-2 selective inhibition often reveal two or more steps in the inhibition process. The overall protein conformations in the crystal structures of COX-1 and COX-2 are not substantially altered upon ligand binding, regardless of ligand type. Thus, the protein dynamics involved in ligand entry may be the critical determinant of ligand binding and binding mode in the COX enzymes.

Citrate bound in the POX site. Stereo view of citrate bound in the POX site of the COX-1·8 structure shown with simulated annealing omit map density contoured to 3σ. Citrate (carbon yellow and oxygen red) straddles the heme (carbon gray, oxygen red, nitrogen blue, and iron rust) and Lys-222 (carbon cyan, nitrogen blue). One carboxylate group of citrate interacts with nitrogen group of Lys-222, whereas another carboxylate group of citrate interacts with a propionate group of heme. In addition, the hydroxyl group of citrate interacts with the propionate group of the heme. Residues proposed to interact with PGG2, the substrate of the peroxidase, are shown as spheres. Site of trypsin cleavage is near residue Arg-277.
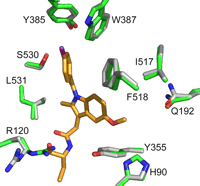
Comparison of the opening of the COX active site. 8 (orange) bound in the COX active site after the structures of COX-1 complexes were superimposed. The green residues represent the positions observed in the COX-1·9 structure, and the gray residues show their positions in the COX-1·8 structure. The only residue to show a significantly altered conformation is Arg-120; in the COX-1·9 complex Arg-120 is extended, whereas it is kinked in the COX-1·8 complex. This conformational change upon the binding of 8 may be the result of some local strain. One consequence of this adjustment of Arg-120 conformation is that the mouth of the active site becomes wider, suggesting that 8 may have a faster dissociation rate as compared with 9.
Anomalous Ligand Interactions in the COX-1·8 Structure—During inspection of the COX-1·8 structure, significant Fo - Fc difference electron density (contoured to 4σ) was observed near the heme within the peroxidase active site. The shape of the density appeared to resemble citrate, which is present in the crystallization buffer. After further refinement with citrate in the model, simulated annealing omit maps yielded defined difference electron density around the citrate molecule (Fig. 4). Citrate is bound with one of its carboxylate groups interacting with one of the propionate groups of the heme. A second interaction involves the amino group of Lys-222 interacting with another one of the carboxylate groups of citrate (i.e. citrate straddles the heme and Lys-222) (Fig. 4). An additional hydrophilic interaction occurs with the hydroxyl group of citrate and the same propionate group of the heme. The potential relevance of citrate binding to the peroxidase active site is that Lys-222 is located in the immediate vicinity of several residues (Lys-211, Glu-289, and Val-291) proposed to interact and possibly stabilize the binding of PGG2, the substrate of the peroxidase reaction (22, 23) (Fig. 4).
What makes the observation of citrate binding in the COX-1·8 structure interesting is that it was not observed in the COX-1·9 structure, despite the fact that the crystallization conditions and crystal handling protocols were identical in both cases. Thus, there must be subtle structural differences induced by inhibitor binding, which favor binding of citrate in one case and not the other. The superposition of several crystal structures of different COX-1-ligand complexes (including NSAIDs and fatty acid substrates) onto the COX-1·9 and COX-1·8 structures reveals that the active site residues displayed only small, and not statistically significant, changes in conformations. However, in the COX-1·8 structure, Arg-120 adopts a significantly altered conformation to accommodate the binding of 8 (Fig. 5). Unlike the more extended conformation seen in the COX-1·9 structure, and in most other COX-1 crystal structures, Arg-120 is found in a more kinked configuration in the COX-1·8 structure. This structural readjustment of Arg-120 results in a slight widening of the mouth to the COX active site suggesting that the binding of 8 may cause local strain, which may, in turn, be manifested in a faster dissociation rate for 8.
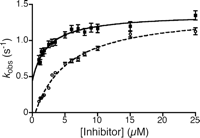
Pre-steady state binding kinetics of compounds 8 and 9 to COX-1. The observed rate constants, kobs of 8 (closed squares) and 9 (open circles) were determined by stopped-flow analysis as described under “Experimental Procedures,” and the dependence of kobs on inhibitor concentration was plotted. The data were fit by nonlinear regression to Equation 2.
The observation of citrate binding in the POX site in the COX-1·8 structure, but not in the COX-1·9 structure, makes the altered conformation of Arg-120 more intriguing. Previous studies have shown that inhibitor binding at the COX site can help stabilize POX activity (24). Other studies have shown that COX inhibitors can also protect the COX-1 enzyme from trypsin cleavage at a surface loop that is near the POX site (25, 26) (Fig. 4), but ∼36 Å away from the COX site. In fact, trypsin cleavage experiments revealed that the extent of proteolysis is ligand-dependent, suggesting that different ligands may induce different degrees of conformation change upon binding, which then result in varying sensitivities to protease cleavage (27). We speculate that the strained binding of a weak COX-1 inhibitor, like 8, promotes citrate binding in the POX active site, whereas the tighter binding mode of 9 disfavors citrate binding. However, the mechanism for transmitting the subtle structural differences resulting from ligand binding to the COX active site to the POX site remains unknown.
Pre-steady State Kinetics of 8 and 9 Binding to COX-1—Potent NSAIDs such as indomethacin are classified as slow, tight-binding inhibitors because they demonstrate time dependence and pseudo-irreversibility of COX inhibition. However, weak COX inhibitors, such as ibuprofen, are typically fast, reversible inhibitors. Analysis of the crystal structures revealed different modes of interaction of 8 and 9 with COX-1, which might be manifested as differences in the observed kinetics of inhibitor binding or dissociation from the enzyme. To probe for differences, the binding kinetics of 8 and 9 were studied.
Many slow, tight binding inhibitors are able to quench the intrinsic tryptophan fluorescence of COX in a time- and concentration-dependent
manner (29). Pre-steady state kinetic measurements of intrinsic protein fluorescence were determined for 8 and 9 in the absence of substrate. Data for each concentration of 8 and 9 were fit to a single exponential decay to extract amplitudes and observed rate constants, kobs. Inhibitor concentration was varied from 5 to 125 times the protein concentration. The amplitudes of the single exponential
decay at concentrations below 2 μm (10× protein concentration) were similar to those above 2 μm, demonstrating binding site saturation at all inhibitor concentrations and validating the use of lower concentrations of
inhibitor for these studies. For both inhibitors, the values for kobs increased with concentration until saturation was achieved (Fig. 6). This is indicative of a multistep mechanism, in which an initial rapid and reversible step is followed by one or more slow
steps (Equation 1), 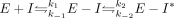
By using this two-step model, kobs was plotted against inhibitor concentration and fit by nonlinear regression to Equation 2, 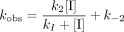
where KI is defined as shown in Equation 3, 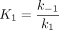
The kinetic constants determined for 8 and 9 are reported in Table 3 and can be compared with values reported for indomethacin that were determined using a similar method (30). KI′ values are comparable for 8 and 9 and are similar to that reported for indomethacin. Furthermore, k2 for 8 and 9 are also comparable, although the rate constant, respectively, is 6 and 10 times faster than that for indomethacin. Notably, whereas 8 was readily reversible with k-2 about half as fast as k2, no reverse rate could be determined for the second step for 9, an effect that is also observed with indomethacin. This difference in k-2 indicates a substantial difference in the tightness of binding of 8 and 9 to COX-1, which correlates to the stereoselectivity of inhibition. Differences in the rate constants for dissociation of COX-inhibitor complexes have been demonstrated previously to be responsible for the COX-2 selectivity of the diarylheterocycle class of inhibitors (31).
Kinetic constants for inhibitor binding
Kinetic constants ± S.E. were determined from secondary plots of kobs determined by stopped-flow analysis as outlined under “Experimental Procedures.”
Time Dependence and Reversibility of COX-1 Inhibition by 8 and 9—To determine whether the differences in the dissociation kinetics of 8 and 9 were correlated to reversibility of inhibition, the time dependence and reversibility of inhibition were investigated. Time dependence of inhibition was examined at a concentration that was five times the IC50 reported for 9 (1.75 μm) and at 75 μm 8, the solubility limit of the compound. The time of preincubation with COX-1 and inhibitor was varied, and the activity was assayed. Notably, both 8 and 9 exhibited minimal time dependence, with maximal inhibition being reached by 30 s. In contrast, the parent compound, indomethacin, requires more than 10 min to achieve maximal inhibition (28). To assess reversibility, COX-1 was preincubated with 50 μm 8 or 9, and the recovery of enzyme activity was assessed following prolonged incubation with arachidonic acid (Fig. 7). For 8, an immediate initial recovery of activity was observed at short time points (less than 45 s), with almost full recovery of activity (92% of control) over 5 min. In contrast, COX-1 preincubated with 9 exhibited only a modest increase (12%) in activity over 5 min. The results of time dependence and reversibility provide a link between inhibitor binding and enzyme inhibition and provide further support that reversibility of binding is the key contribution to the kinetic basis of enantioselectivity of α-substituted indomethacin ethanolamides.
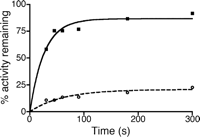
Reversibility of COX-1 inhibition by compounds 8 and 9. COX-1 was preincubated with 50 μm 8 (closed squares) or 9 (open circles) prior to addition of arachidonic acid. Aliquots were removed at various times (30 s to 5 min) after addition of arachidonic acid, and the conversion of arachidonic acid to oxygenated products was quantified. Activity was normalized to Me2SO controls and plotted against the time of incubation with arachidonic acid. Arachidonic acid is able to efficiently compete with 8 (closed squares) for COX-1 active site, but only modest recovery of activity is observed for 9 (open circles).
Summary—The COX-1 crystal structures and binding studies with 8 and 9 provide important insights into the physical and kinetic parameters that define the modes of binding and chiral selectivity of these α-substituted indomethacin ethanolamides. Several unanswered questions still remain. (a) Why do the S-α-substituted ethanolamides with quite bulky substituents inhibit just as well as the S-stereoisomers with less bulky substituents? (b) How bulky an α-substitution can the side pocket in COX-1 accommodate? (c) Would the binding mode of the S-α-substituted compounds shift if the substitution is too large? The crystal structure of the COX-1·9 complex clearly shows that there is enough space for the ethyl group, but this may not be the case for larger moieties.
A recent modeling study (32) investigated the basis of stereoselective binding using several indomethacin ethanolamides possessing the larger isopropyl substituent. Although the binding modes obtained for the S-series of inhibitors were not similar to that seen in the COX-1·9 structure, they did reveal that the inhibitor having the (R)-isopropyl group could achieve the same binding interactions as the S-enantiomer but at the cost of maintaining a constrained conformation. This directly supports what was observed in the crystal structure in that the binding of 8 induces strain within the protein, as evidenced by altered conformation of Arg-120.
However, the physical basis for the chiral selectivity of COX-1 for a wider range of α-substitutions may turn out to be much more complicated than simple steric hindrance. For example, the strong interactions between the ethanolamide group of 9 compensates for the lack of a strong interaction between a carboxylate group and Arg-120 and Tyr-355, which is necessary for high affinity binding of classical NSAIDs to COX-1. Crystal structures of COX-1 complexed with α-substituted indomethacin ethanolamides having larger α-substitutions may provide further information about interactions that define the different binding modes within the COX active site.
Acknowledgments
All data presented here were collected at COM-CAT (Sector 32 beamline) at Advanced Photon Sources (Argonne, IL), and we thank Joe Brunzelle of LS-CAT for assistance during our time at COM-CAT. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. LS-CAT is supported in part by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor Grant 085P1000817.
Footnotes
-
↵ 4 The abbreviations used are: COX, cyclooxygenase; PG, prostaglandin; POX, peroxidase; NSAIDs, nonsteroidal anti-inflammatory drugs; β-OG, n-octyl-β-glucopyranoside; oCOX, ovine COX.
-
↵ 5 The numbering of residues in ovine COX-1 is based on numbering the N-terminal methionine of the signal peptide as residue number 1.
-
↵ 6 The designations 8 and 9 derive from the paper describing their synthesis (15).
-
The atomic coordinates and structure factors (code 2OYE and 2OYU) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
-
↵* This work was supported in part by National Institutes of Health Grants R01HL56773, P01GM57323, R01GM68868, and R01CA89450 and National Institutes of Health Training Grant T32ES07028 (to M. V. T.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
-
↵1 Both authors contributed equally to this work.
-
↵2 Recipient of Ruth L. Kirschstein National Research Service Award F31DA02014 from the National Institute on Drug Abuse and a fellowship from the Vanderbilt Institute of Chemical Biology.
- Received February 15, 2007.
- Revision received June 26, 2007.
- The American Society for Biochemistry and Molecular Biology, Inc.












