The cyclooxygenases (COX-1 and COX-2) generate prostaglandin H2 from arachidonic acid (AA). In its catalytically productive conformation, AA binds within the cyclooxygenase channel with its carboxylate near Arg-120 and Tyr-355 and ω-end located within a hydrophobic groove above Ser-530. Although AA is the preferred substrate for both isoforms, COX-2 can oxygenate a broad spectrum of substrates. Mutational analyses have established that an interaction of the carboxylate of AA with Arg-120 is required for high affinity binding by COX-1 but not COX-2, suggesting that hydrophobic interactions between the ω-end of substrates and cyclooxygenase channel residues play a significant role in COX-2-mediated oxygenation. We used structure-function analyses to investigate the role that Arg-120 and residues lining the hydrophobic groove play in the binding and oxygenation of substrates by murine (mu) COX-2. Mutations to individual amino acids within the hydrophobic groove exhibited decreased rates of oxygenation toward AA with little effect on binding. R120A muCOX-2 oxygenated 18-carbon ω-6 and ω-3 substrates albeit at reduced rates, indicating that an interaction with Arg-120 is not required for catalysis. Structural determinations of Co3+-protoporphyrin IX-reconstituted muCOX-2 with α-linolenic acid and G533V muCOX-2 with AA indicate that proper bisallylic carbon alignment is the major determinant for efficient substrate oxygenation by COX-2. Overall, these findings implicate Arg-120 and hydrophobic groove residues as determinants that govern proper alignment of the bisallylic carbon below Tyr-385 for catalysis in COX-2 and confirm nuances between COX isoforms that explain substrate promiscuity.
Prostaglandins are fatty acid-derived lipid mediators involved in the regulation of various homeostatic functions required for normal physiological activities as well as central players in a number of pathologies, including pain, inflammation, and cancer. These potent signaling molecules are generated from prostaglandin H2 (PGH2),3 which is produced via the oxygenation of arachidonic acid (AA; 20:4 ω-6) by the cyclooxygenase enzymes (COX-1 and COX-2) (1). COX-1 and COX-2 are the targets of aspirin and other nonsteroidal anti-inflammatory drugs utilized in the treatment of pain and inflammation (2). Diaryl heterocycle-based compounds, including rofecoxib and celecoxib, selectively inhibit COX-2 (3).
COX-1 and COX-2 are heme-containing sequence homodimers that associate with one leaflet of the membrane bilayer. Each monomer of the homodimer contains a spatially distinct peroxidase and cyclooxygenase active site, which are functionally linked via the heme moiety. Although the sequence of each monomer of the homodimer is identical, the enzymes exhibit half-of-sites activity such that at a given time only one monomer of the homodimer is catalytically active (4,–,6). COX-1 displays a limited fatty acid substrate specificity, preferentially oxygenating AA. Conversely, COX-2 efficiently oxygenates a broad spectrum of fatty acid and ester substrates, including AA, eicosapentaenoic acid (EPA; 20:5 ω-3), docosahexaenoic acid (22:6 ω-3), the essential fatty acids linoleic acid (LA; 18:2 ω-6) and α-linolenic acid (αLA; 18:3 ω-3), and the endocannabinoids 2-arachidonoyl glycerol and anandamide (6,–,10). The activity of the “catalytic” monomer can be modulated by nonsubstrate fatty acids, such as palmitic acid (PA; 16:0), which binds in the cyclooxygenase channel of the opposite monomer, termed the “allosteric” monomer (4).
Crystal structures of COX-1 and COX-2 in complex with AA and other substrates provide a detailed picture at the molecular level of the interactions between substrate and active site residues necessary for binding and subsequent catalysis (11,–,15). AA binds in its catalytically productive conformation within the cyclooxygenase channel with its carboxylate located near the side chains of Arg-120 and Tyr-355 at the opening of the channel and with its ω-end located in a hydrophobic groove above the side chain of Ser-530 such that carbon 20 of AA lies adjacent to Gly-533 (see Fig. 1). In this configuration, carbon 13 is placed ∼3.0 Å below the phenolic oxygen of Tyr-385, which is poised for hydrogen abstraction and subsequent initiation of cyclooxygenase catalysis. The crystal structures of COX-1 and COX-2 are virtually superimposable. As such, AA and other substrates have been observed to bind productively within the cyclooxygenase channel in globally similar L-shaped conformations with the contacts made between substrate and residues lining the channel predominantly conserved (12,–,15).
There are structural nuances between COX-1 and COX-2 that result in an increase in substrate promiscuity and selective inhibition of COX-2. Hallmark among these differences are the substitutions of cyclooxygenase channel residues Ile-434, His-513, and Ile-523 in COX-1 to Val-434, Arg-513, and Val-523 in COX-2, resulting in a ∼25% increase in the volume of the cyclooxygenase channel and the formation of an isoform-specific side pocket (16). This side pocket with Arg-513 located at its base has been exploited in the design of inhibitors aimed at reducing toxic gastrointestinal side effects caused by conventional nonsteroidal anti-inflammatory drugs (3). A second nuance is the observed flexibility of the side chain of Leu-531 in COX-2 that is located at the mouth of the cyclooxygenase channel above the side chain of Arg-120. The increased flexibility of the Leu-531 side chain increases the volume of the cyclooxygenase channel of COX-2 and permits the binding of endocannabinoid substrates (14, 15). A third nuance is the requirement of an ionic interaction between the carboxylate group of AA and the side chain of Arg-120 for high affinity binding of AA to COX-1 but not COX-2 (15, 17). Studies utilizing a glutamine substitution of Arg-120 showed a greater than 1000-fold increase in the Km value of AA for COX-1 (18) but little to no change for the equivalent substitution in COX-2 (17). A final nuance is the variable sensitivity of each isoform to mutation at Gly-533. Although a G533A substitution of COX-1 has no detectable cyclooxygenase activity (19), the same substitution in COX-2 results in a mutant construct that generates prostaglandin products albeit with diminished specific activity (20).
The x-ray crystal structures of AA in complex with murine (mu) COX-2 and ovine (ov) COX-1 illustrate that carbons 13–20 of AA contribute to 40 of the 54 observed contacts (74%) between substrate and cyclooxygenase channel residues in COX-2 compared with 26 of the 52 contacts (50%) observed in COX-1 (11, 15). This fact along with the Arg-120 and Gly-533 mutagenesis studies suggests that hydrophobic contacts at the ω-end of the substrate play a more prominent role in high affinity binding of AA to COX-2 (17). Here we report studies designed to further investigate the role that the residues lining the hydrophobic groove above Ser-530 have in the binding and oxygenation of AA and other 18–20-carbon ω-6 and ω-3 fatty acid substrates. Our functional studies reveal that residues occupying the hydrophobic groove do not individually influence substrate binding affinity but that these side chains along with Gly-533 and Arg-120 are important for proper pro-S hydrogen positioning below Tyr-385 for efficient catalysis in COX-2. X-ray crystallographic analysis of a G533V muCOX-2 construct in complex with AA confirms this observation, whereas the crystal structure of αLA bound to wild type muCOX-2 elucidates for the first time how 18-carbon fatty acids bind to COX-2.
The fatty acids AA (5Z,8Z,11Z,14Z-eicosatetraenoic acid), LA (9Z,12Z-octadecadienoic acid), αLA (9Z,12Z,15Z-octadecatrienoic acid), stearidonic acid (SA; 6Z,9Z,12Z,15Z-octadecatetraenoic acid), EPA (5Z,8Z,11Z,14Z,17Z-eicosapentaenoic acid), and PA (hexadecanoic acid) were purchased from Cayman Chemical Co. (Ann Arbor, MI). Fe3+- and Co3+-protoporphyrin IX were purchased from Frontier Scientific (Logan, UT). Decyl maltoside and n-octyl β-d-glucopyranoside (βOG) were purchased from Anatrace (Maumee, OH). The QuikChange™ mutagenesis kit was purchased from Stratagene (La Jolla, CA). SDS-PAGE and Novex® NativePAGE™ Bis-Tris Gel System components as well as the Bac-to-Bac® baculovirus expression kit and associated reagents, including Spodoptera frugiperda 21 (Sf21) insect cells, fetal bovine serum, Fungizone, penicillin-streptomycin, and sf-900 III serum-free medium were purchased from Invitrogen. HiTrap™ HP Chelating and HiPrep™ Sephacryl S300-HR chromatography columns were purchased from Amersham Biosciences. Oligos used for site-directed mutagenesis were purchased from Integrated DNA Technologies (Coralville, IA).
Single mutant constructs and the double mutant R120A/G533A construct were created with the QuikChange mutagenesis kit using the primers listed in supplemental Table S1. For the single mutant constructs, we used His6-N580A muCOX-2 in pFastBac1 as the template (15), whereas the R120A/G533A double mutant was created by using the R120A mutant construct as the template. Each mutant construct was verified by DNA sequence analysis at the Roswell Park Cancer Institute DNA Sequencing Laboratory. The expression and purification of muCOX-2 and associated mutants were carried out as described (15). For functional studies, wild type and mutant constructs were subjected to immobilized metal affinity chromatography only. For crystallization, immobilized metal affinity chromatography-purified proteins were trypsin-digested for 20–90 min at 25 °C with a 30:1 ratio of COX-2 to protease followed by termination of the reaction via the addition of 2 mm phenylmethylsulfonyl fluoride. Trypsinized protein was then subjected to size exclusion chromatography utilizing a HiPrep 16/60 Sephacryl S-300 HR column equilibrated in 25 mm Tris, pH 8.0, 150 mm NaCl, and 0.53% (w/v) βOG. Peak fractions were pooled and concentrated to 3 mg/ml for crystallization trials. Cyclooxygenase and peroxidase activity were measured as described (15).
For crystallization, purified apoproteins were reconstituted with a 2-fold moles excess of Co3+-protoporphyrin IX and dialyzed overnight at 4 °C against 20 mm Tris, pH 8.0, 100 mm NaCl, and 0.6% (w/v) βOG. Subsequently, a 10-fold moles excess of fatty acid was added to the reconstituted protein to generate the muCOX-2·αLA and G533V·AA complexes. Crystals were grown using the sitting drop vapor diffusion technique by mixing 3 μl of protein solution with 3 μl of a solution consisting of 23–34% polyacrylic acid 5100, 100 mm HEPES, pH 7.5, 20 mm MgCl2, and 0.6% (w/v) βOG. Drops were then equilibrated over reservoir solutions containing 23–34% polyacrylic acid 5100, 100 mm HEPES, pH 7.5, and 20 mm MgCl2 at 23 °C. Crystals were cryoprotected using 15% ethylene glycol as described (15) and flash cooled directly in the nitrogen gas stream prior to diffraction analysis. Data were collected on beamline A1 at the Cornell High Energy Synchrotron Source using an Area Detector Systems CCD Quantum-210 detector. Data sets were integrated and scaled using MOSFLM and SCALA, respectively, in the CCP4 suite of programs (21). Details of the data collection statistics are summarized in Table 1.
Crystallographic statistics for the G533V·AA and muCOX-2·αLA crystal structures
Molecular replacement methods were used to determine initial phases for each structure. As detailed in Vecchio et al. (15), efforts were made to reduce the introduction of model bias through the use of a truncated search model of muCOX-2 in Phaser (22) followed by input of the phases from the molecular replacement solution for each model into ARP/wARP utilizing the “automated model building starting from experimental phases” option (23). ARP/wARP built most of the deleted portions of the models. Iterative cycles of manual model building and refinement using Coot (24) and REFMAC5 (25) were carried out to fit the remaining residues and to add waters, substrate, and other ligand molecules. TLS refinement (26) utilizing the TLSMD web server (27, 28) was carried out during the final rounds of refinement. The final models for each crystal structure of muCOX-2 consist of residues 33–583 for monomer A and 33–584 for monomer B, Co3+-protoporphyrin IX, sugar moieties, water molecules, cryoprotectant molecules, and one βOG molecule along with the appropriate fatty acid substrate. Final refinement statistics are summarized in Table 1.
To further validate the observed conformation of AA in the G533V·AA crystal structure, we modeled AA upside down with its carboxylate tethered to the side chains of Tyr-385 and Ser-530 (as observed in monomer A of Protein Data Bank code 3HS5 (15)). Both 2Fo − Fc and Fo − Fc electron density maps were calculated after 25 cycles of restrained refinement in REFMAC5. The presence of negative electron density over the carboxylate group and positive electron density over the ω-end of AA clearly indicated that the substrate binds with its carboxylate group near the side chain of Leu-531 at the opening of the cyclooxygenase channel (data not shown).
For structural analyses, van der Waals and hydrogen bond interactions were calculated using the program CCP4MG (29). The upper limit on distance for consideration as a van der Waals contact was 4.0 Å. Superposition of coordinates between structures was done using the program LSQAB within the CCP4 suite of programs and the coordinates for all Cα atoms unless otherwise stated. Simulated annealing omit maps were calculated using CNS (30). Model validation and Ramachandran plot analyses were carried out using MolProbity (31). Figures were created using CCP4MG (29). The coordinates and structure factors for G533V·AA and muCOX-2·αLA have been deposited in the Protein Data Bank (Protein Data Bank codes 3TZI and 4E1G, respectively).
Paramount to the productive binding, alignment, and subsequent oxygenation of AA and other fatty acid substrates by COX-1 and COX-2 is the insertion of the ω-end of the substrate into the hydrophobic groove located above the side chain of Ser-530 at the apex of the cyclooxygenase channel (11,–,15). To probe the role that residues lining the hydrophobic groove play in high affinity binding and oxygenation of AA to COX-2, we generated mutant constructs of Phe-205, Phe-209, Ile-377, Phe-381, and Gly-533. These residues participate in 25 hydrophobic contacts with carbons 14–20 of AA in wild type COX-2 (Fig. 1) (15). Substitutions of alanine, valine, leucine, or phenylalanine (Ile-377 only) were made followed by expression in insect cells and purification utilizing immobilized metal affinity chromatography. All mutant constructs exhibited peroxidase activity with the exception of I377A, I377F, F381A, F381V, and F381L. These five mutants also exhibited altered electrophoretic mobility compared with wild type enzyme on a native gel (data not shown), suggesting that these substitutions result in the production of improperly folded enzyme. The purity of wild type and each functional mutant construct was assessed to be greater than 85% after the immobilized metal affinity chromatography step using SDS-PAGE (data not shown). Table 2 summarizes the kinetic parameters for each mutant construct utilizing AA as the substrate as well as the relative peroxidase rates of each mutant compared with wild type enzyme.
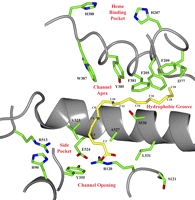
AA bound in the cyclooxygenase channel of wild type muCOX-2. A view of the wild type muCOX-2 cyclooxygenase active site and corresponding residues probed in this study is shown. The structure depicts Protein Data Bank code 3HS5 (muCOX-2·AA) monomer B with AA (yellow carbons) bound productively. Every other carbon of AA is labeled for clarity, and muCOX-2 amino acids are labeled according to the traditional ovCOX-1 numbering scheme.
Kinetic parameters generated for wild type and mutant muCOX-2 constructs using AA
kcat and Km values were derived from three independent determinations (±S.E.) using 24–72 nm protein and an oxygen electrode. Efficiency (E) is defined as the kcat/Km. Values for the relative peroxidase activity represent the average of two measurements followed by normalization to the rate of wild type enzyme. ND, not determined.
Phe-205 makes four hydrophobic contacts with carbons 14–16 and one to carbon 18 of AA within the hydrophobic groove (15). Substitution of Phe-205 with alanine resulted in a greater than 70% reduction in relative cyclooxygenase activity, whereas substitutions with valine and leucine had more moderate effects when compared with wild type enzyme. The enzyme efficiencies of all Phe-205 mutants were decreased between 2.7- and 4.6-fold compared with wild type enzyme. Phe-209 makes 10 contacts with carbon 16 and carbons 18–20 of AA. Only substitution of alanine at this position resulted in a reduction of cyclooxygenase activity and catalytic efficiency. Hence, for Phe-205 and -209, the more conservative substitutions of valine and leucine, which would provide an increase in volume of the hydrophobic groove while maintaining the potential for the formation of stabilizing hydrophobic contacts, afford AA a higher degree of conformational freedom at the ω-end to position carbon 13 optimally below Tyr-385 for catalysis. None of the Phe-205 or Phe-209 substitutions led to increases greater than 1.8-fold in Km for AA compared with wild type enzyme, indicating that other residues lining the hydrophobic groove can compensate for the lost contacts with AA. The observed kinetic behaviors for substitutions at Phe-205 and Phe-209 are also in line with equivalent characterizations carried out using ovCOX-1 (32).
Ile-377 makes a single hydrophobic contact with carbon 20 when AA is bound productively in COX-1 and COX-2 (11, 15). Conservative substitutions of valine and leucine at this position in muCOX-2 resulted in only a modest decrease in the oxygenation of AA compared with wild type enzyme, which is similar to that observed for the equivalent substitution in ovCOX-1 (32). Furthermore, there were no significant changes in Km values or efficiencies for these mutant constructs. Interestingly, alanine and phenylalanine substitutions resulted in misfolded proteins, indicating that a significant increase or decrease in side chain length at this position not only compromises the structural integrity of the hydrophobic groove but also the enzyme as a whole. Gly-533 makes five hydrophobic contacts via its main chain Cα and carbonyl carbon atoms with carbons 17–19 of AA. The ω-end of AA lies above Gly-533 just as it abuts against the side chain of Ile-377. Consistent with previous studies, a G533A mutant retained only 7% cyclooxygenase activity compared with wild type enzyme, whereas G533V and G533L substitutions completely abolished cyclooxygenase activity (5, 9).
In addition to probing hydrophobic pocket residues, we constructed a mutant of Arg-120 as well as an R120A/G533A double mutant. Arg-120 does not make any contacts to AA in its productive conformation in the muCOX-2·AA crystal structure (Fig. 1) (15). Substitution of Arg-120 with alanine had no effect on oxygenation of AA but did increase the Km 3.4-fold compared with wild type enzyme (Table 2). By contrast, the R120A/G533A double mutant did not retain any cyclooxygenase activity.
Taken together, our mutational and kinetic analyses indicate that Phe-205, Phe-209, and Ile-377 are not individually responsible for high affinity binding of AA within the cyclooxygenase channel of muCOX-2 given that Km values for substitutions at these positions are only modestly increased compared with wild type enzyme. Instead of stabilizing the ω-end of AA within the hydrophobic groove, we speculate that these residues contribute to the proper alignment of carbon 13 below Tyr-385 for optimal catalysis. Substitutions I377V, I377L, and G533A, which reduce the volume of the hydrophobic groove, have a similar effect in that they cause a misalignment of carbon 13 below Tyr-385 and hence a decrease in activity compared with wild type enzyme. Finally, our functional analysis of the R120A muCOX-2 construct was consistent with previous studies that indicated a lack of involvement of the side chain of Arg-120 in high affinity binding of AA to COX-2 (17).
We utilized x-ray crystallography to validate the hypothesis that the reduced rates of AA oxygenation by Gly-533 mutants are due to the misalignment of carbon 13 below Tyr-385 rather than an inability of substrate to bind within the cyclooxygenase channel. A complex of AA bound to G533V muCOX-2 was generated by incubating the substrate with Co3+-protoporphyrin IX-reconstituted mutant enzyme followed by crystallization and subsequent diffraction analysis using synchrotron radiation. Crystals of the complex diffracted to 2.15 Å and revealed the characteristic domain features expected of COX-1 and COX-2 crystal structures, including the epidermal growth factor-like domain, membrane binding domain, and catalytic domain (Table 1). The two monomers that comprise the asymmetric unit of the G533V·AA crystal structure are virtually identical given the calculated root mean square difference between monomers of 0.19 Å (for 550 Cα pairs).
Clear electron density was observed for AA within the cyclooxygenase channel of each monomer of the G533V·AA crystal structure (Fig. 2, A and B). In both monomers, AA adopts an overall “C-shaped” conformation, which is novel compared with previously observed conformations of AA and other fatty acid substrates bound to COX-1 and COX-2 (11,–,13, 15). Because the ω-end of AA cannot fully penetrate the hydrophobic groove, AA is shifted within the channel, subsequently forcing the side chain of Leu-531 to adopt an alternate rotamer conformation to accommodate the carboxylate end of AA (Fig. 2, A and B). The movement of the Leu-531 side chain is identical to that observed when the endocannabinoid substrate 1-arachidonoyl glycerol binds within the cyclooxygenase channel of muCOX-2 (14). The resulting shift of AA within the channel places the carboxylate greater than 6.5 Å away from the side chain of Arg-120 in each monomer. Interestingly, the C-shaped binding mode mimics the nonproductive binding mode of AA observed in monomer A of the muCOX-2·AA crystal structure (15, 33) except that the carboxylate of AA is bound near the opening of the channel instead of near Tyr-385 and Ser-530 at the channel apex (Fig. 2C).
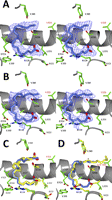
AA bound in the cyclooxygenase channel of G533V muCOX-2. Stereoviews of the C-shaped nonproductive conformations of AA (blue carbon and red oxygen atoms) bound in the cyclooxygenase channel of monomer A (A) and monomer B (B) of the G533V·AA crystal structure are shown. The Fo − Fc simulated annealing omit electron density contoured at 4σ is shown with the final refined models of AA. C, comparison of the nonproductive binding conformations of AA observed within the cyclooxygenase channel of the crystal structures of G533V·AA monomer B (blue AA and green side chains) versus muCOX2·AA monomer A (yellow AA and yellow side chains). Alternate positions of the C8-C9 double bond are observed for the nonproductive conformations of AA bound in monomer A of muCOX-2·AA. D, comparison of the nonproductive conformation of AA observed within monomer B of G533V·AA and the productive conformation of AA observed within monomer B of muCOX2·AA colored as described in C. Carbon 13 positions of AA in the wild type and G533V crystal structures are labeled with black and red asterisks, respectively. In all panels, side chain atoms of residues lining the cyclooxygenase channel are labeled accordingly with the valine mutation at position 533 labeled in red.
The observed C-shaped conformation of AA fully explains why the G533V substitution results in the complete abolishment of cyclooxygenase activity due to the misalignment of carbon 13 below the phenolic oxygen of Tyr-385. Carbon 13 is displaced by greater than 6 Å compared with its position when AA is bound productively within the cyclooxygenase channel (Fig. 2D). AA makes only 29 contacts with 14 residues in monomer A and 25 contacts with 12 residues in monomer B (supplemental Table S2) compared with 52 contacts with 19 residues when bound productively to wild type enzyme (15). The lack of deep penetration of the ω-end of AA into the hydrophobic groove results in the loss of 20 contacts with the side chains of Phe-209, Ile-377, Phe-381, and Gly-533. Only the side chain of Phe-205 makes a single contact with AA in its C-shaped conformation, but even this represents a net loss as Phe-205 makes five contacts to AA when it is bound productively within the channel (15). Thus, our crystallographic analysis supports our functional studies and confirms that AA binds to Gly-533 mutants of muCOX-2. Substitution of valine for Gly-533 adds steric bulk to the hydrophobic groove, resulting in the binding of AA in a novel, nonproductive conformation due to the lack of penetration into the hydrophobic groove.
We next focused our efforts on the proximal (Arg-120) and distal (Gly-533) extremes of the cyclooxygenase channel given that individual mutations to hydrophobic groove residues showed no marked effect on AA binding to COX-2. Utilizing alternate fatty acid substrates possessing varying chain lengths and degrees of unsaturation as tools, we sought to better understand how Arg-120 and Gly-533 influence the binding and oxygenation of substrates by COX-2.
The 18-carbon fatty acid substrates αLA (18:3 ω-3) and SA (18:4 ω-3) are unique substrates for wild type muCOX-2 in that they possess allylic carbons at both the ω-8 and ω-5 positions that can be oxygenated by COX-2 (7, 9). Previous mutational studies showed that G533A, G533V, and G533L muCOX-2 mutants oxygenate SA and αLA to varying degrees (9). Presumably, the Gly-533 substitutions restrict full penetration of the ω-ends of these substrates into the hydrophobic groove, resulting in the alignment of the ω-5 allylic carbon below Tyr-385 for hydrogen abstraction and subsequent catalysis. Given this premise, we hypothesize that the binding determinant(s) for SA and αLA is located near the opening of the cyclooxygenase channel with potential involvement of the side chain of Arg-120. Lending support to this hypothesis is the recent structural characterization of the non-substrate fatty acid PA (16:0) bound within the cyclooxygenase channel of muCOX-2 (4). PA, which modulates the activity of COX-2 through its binding to the allosteric monomer of the COX-2 homodimer, is four carbons shorter than AA and does not penetrate the hydrophobic groove when bound within the channel. As such, there are no interactions between PA and the side chains of Phe-209, Ile-377, Phe-381, and Gly-533. To compensate for these lost contacts, there is an increase in the number of ionic and hydrogen bonds formed between the carboxylate of PA and the side chain atoms of Arg-120 and Tyr-355 at the opening of the channel.
The specific cyclooxygenase activities of wild type, R120A, G533A, G533V, and the R120A/G533A double mutant construct were measured utilizing AA, αLA, and SA as well as two additional fatty acid substrates, EPA and LA (supplemental Fig. S1). EPA is a 20-carbon substrate that possesses both ω-8 and ω-5 allylic carbons, whereas LA is an 18-carbon substrate that contains only an ω-8 allylic carbon. Both have been previously characterized as COX-2 substrates (6, 7). In addition, kinetic parameters were determined for the G533A mutant construct with LA, αLA, SA, and EPA.
In agreement with previously reported studies, wild type muCOX-2 oxygenated LA, αLA, SA, and EPA at rates 25–40% of the oxygenation rate of AA (Fig. 3) (6, 7, 9). Km values calculated for LA, αLA, SA, and EPA using the wild type construct were 2.2-, 4.6-, 1.8-, and 1.1-fold higher than that of AA in line with values reported previously for LA and αLA with human COX-2 (Table 3) (7). The reduced oxygenation rates of these substrates coupled with low binding affinities resulted in enzyme efficiencies between 3.6- and 8-fold lower than that exhibited by AA. G533A muCOX-2 effectively oxygenated all ω-3 fatty acid substrates at rates equivalent to wild type enzyme, whereas ω-6 fatty acids were oxygenated at significantly diminished rates by comparison (Fig. 3). G533A muCOX-2 had Km values for αLA and SA that were decreased ∼1.4-fold compared with wild type enzyme, whereas the Km for EPA increased by the same magnitude (Table 3). The G533V mutant construct did not oxygenate AA or LA and exhibited a greater than 95% reduction in the rate of oxygenation when αLA, SA, and EPA were utilized as substrates (Fig. 3).
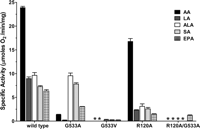
Oxygenation of ω-6 and ω-3 fatty acid substrates by wild type and mutant muCOX-2 constructs. A bar graph depicting the initial rates of oxygenation of ω-6 and ω-3 fatty acids by wild type, G533A, G533V, R120A, and R120A/G533A mutant constructs is shown. Measurements were carried out utilizing 100 μm AA, LA, αLA (ALA), SA, and EPA and corrected where necessary for the percentage of mono- and bisoxygenated products generated. Enzyme concentrations used were 24 nm for wild type, G533A, and R120A constructs and 48 nm for G533V and the R120A/G533A mutant construct. Error bars represent the S.D. between duplicate measurements. *, no detectable activity.
Kinetic constants generated for wild type and G533A muCOX-2 using ω-6 and ω-3 fatty acid substrates
Values were derived from three independent determinations (±S.E.). utilizing an oxygen electrode. Vmax and subsequent kcat values are corrected for the percentage of mono- and bisoxygenated products. Efficiency (E) is defined as the kcat/ Km. ND, not determined.
AA was efficiently oxygenated by R120A muCOX-2, although there was an increase in the Km value (Fig. 3 and Table 2). This result is consistent with earlier mutational and structural studies showing that an ionic interaction between the carboxylate of AA and the side chain of Arg-120 is not required for high affinity binding and subsequent oxygenation of AA (15, 17). LA, αLA, SA, and EPA were also oxygenated by R120A muCOX-2 albeit at reduced rates compared with wild type enzyme (Fig. 3). The more dramatic effect on the oxygenation of these substrates compared with AA suggests that the side chain of Arg-120 plays a more prominent role in the binding and oxygenation of LA, αLA, SA, and EPA within the cyclooxygenase channel for catalysis. The R120A/G533A double mutant was not able to oxygenate AA, LA, αLA, or SA (Fig. 3). Interestingly, EPA was oxygenated by the R120A/G533A double mutant at rates similar to those of the R120A mutant construct alone. We speculate that the additional rigidity and length afforded EPA by the presence of five double bonds within the 20-carbon substrate chain provides enough inherent stabilization within the cyclooxygenase channel to allow for hydrogen abstraction from the ω-5 allylic carbon.
To further delineate the role of Arg-120 in the oxygenation of these substrates, we performed mixed substrate assays in which the oxygenation of AA in the presence of excess 18-carbon substrate was compared with the oxygenation of AA alone using wild type and R120A muCOX-2 (Table 4). In all cases, the presence of an 18-carbon fatty acid reduced the oxygenation of AA by COX-2 between 14 and 22%, indicating that there is competition between substrates for binding within the cyclooxygenase channel. Competition was also observed for the R120A mutant construct with LA and αLA reducing AA oxygenation rates by ∼40% and SA reducing AA oxygenation rates by 24% (Table 4). Based on the R120A competition results, we speculate that the side chain of Arg-120 is critical for the positioning of LA, αLA, and SA for optimal hydrogen abstraction rather than serving as a high affinity binding determinant for substrate within the cyclooxygenase channel.
Effect of PA, LA, αLA, and SA on the oxygenation of AA by wild type and R120A muCOX-2
For the competition assay, results are expressed as the percent decrease in oxygenation of 20 μm AA in the presence of 100 μm 18-carbon fatty acid substrate relative to the oxygenation of 20 μm AA alone. For the activation assay, results are expressed as the percent increase in oxygenation of 20 μm AA in the presence of 100 μm PA relative to the oxygenation of 20 μm AA alone. Values are derived from triplicate determinations using an oxygen electrode (±S.E.).
We also tested whether PA could increase the rate of oxygenation of AA and the other fatty acid substrates utilized in this study. Consistent with studies carried out using human COX-2, PA increased the rate of oxygenation of AA by muCOX-2 by 13% (Table 4) (4, 6). Moreover, PA increased the rate of oxygenation of LA, αLA, and SA by muCOX-2 by 29, 7, and 33%, respectively. Conversely, PA did not potentiate the oxygenation of AA when the R120A construct was utilized (Table 4). This observation is consistent with structural data that identified stabilizing ionic and hydrogen bonds between the side chain of Arg-120 and the carboxylate of PA to be critical for binding of PA within the cyclooxygenase channel (4).
To examine the binding conformation of an 18-carbon ω-3 fatty acid substrate within the cyclooxygenase channel, we determined the x-ray crystal structure of Co3+-protoporphyrin IX-reconstituted muCOX-2 with αLA (muCOX-2·αLA) to 2.1-Å resolution using synchrotron radiation. As observed with the G533V·AA crystal structure, the muCOX-2·αLA complex crystallized with two monomers in the asymmetric unit with the root mean square difference between monomers calculated to be 0.23 Å (for 551 Cα atoms). Clear electron density was observed for αLA within the cyclooxygenase channel of each monomer of the muCOX-2·αLA structure (Fig. 4, A and B). The substrate chains adopt nearly identical conformations in each monomer that are similar to the canonical L-shaped conformation observed for AA and other 20-carbon fatty acid substrates (11,–,13, 15). αLA makes 54 contacts with residues lining the cyclooxygenase channel in monomer A (supplemental Table S3) and 55 contacts in monomer B (supplemental Table S4). There are no significant differences in the enzyme-substrate contacts between monomers of the muCOX-2·αLA structure or with contacts previously identified in other COX-2·fatty acid substrate structures (15). However, it is interesting to note that the O1 atom of the carboxylate of αLA forms two hydrophilic contacts with the Nε and Nζ2 group of Arg-120 at the opening of the channel. Neither the carboxylate of AA nor that of EPA interacts with the side chain of Arg-120 when they are bound productively within the cyclooxygenase channel of COX-2 (15, 17).
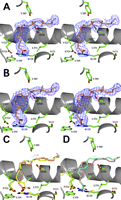
αLA bound in the cyclooxygenase channel of wild type muCOX-2. Stereoviews of the observed conformations of αLA bound within the cyclooxygenase channel of monomer A (A) and monomer B (B) of the muCOX2·αLA crystal structure are shown. The Fo − Fc simulated annealing omit electron density contoured at 4.5σ is shown in blue with the final refined models of αLA depicted with copper carbon atoms and red oxygen atoms. Hydrogen atoms are also shown modeled onto carbon 11 and carbon 14 of αLA. The side chain atoms of residues lining the cyclooxygenase channel are colored green and labeled accordingly. C, comparison of the conformation of AA (yellow) bound within the cyclooxygenase channel of monomer B in the muCOX-2·AA crystal structure and the conformation of αLA (copper) bound to monomer B of muCOX-2·αLA. D, comparison of the conformation of LA (cyan) bound within the cyclooxygenase channel of ovCOX-1 (Protein Data Bank code 1IGZ (12)) and the conformation of αLA (copper) bound to monomer B of muCOX-2·αLA. In C and D, the position of carbon 11 in LA and αLA is depicted by an asterisk colored in cyan, and the position of carbon 13 of AA is depicted by an asterisk colored in red.
The 18-carbon chain of αLA extends into the hydrophobic groove as deeply as 20-carbon substrates and forms an “S-shaped” kink that weaves the central portion of the substrate around the side chain of Ser-530 (Fig. 4C). The variation observed between carbons 5–10 of αLA and AA due to the lack of double bonds in this region of αLA provides the necessary flexibility for extension of the ω-end into the hydrophobic groove (Fig. 4C). In this conformation, the ω-8 allylic carbon of αLA (carbon 11) is positioned ∼3 Å below the phenolic oxygen of Tyr-385 for hydrogen abstraction, analogous to the positioning of carbon 13 of AA and EPA in their productive conformations (15). Conversely, the ω-5 allylic carbon of αLA (carbon 14) is positioned ∼4.5 Å away. The extended conformation observed for αLA is also similar to that seen for LA bound within the cyclooxygenase channel of ovCOX-1 (Fig. 4D) (12). Although αLA contains an additional double bond at its ω-end compared with LA, both substrates occupy the same volume in the hydrophobic groove, and both substrates have carbon 11 positioned optimally below the side chain of Tyr-385 (Fig. 4D).
Previous studies focused on delineating the molecular determinants driving high affinity binding of substrates to COX-2 speculated that hydrophobic contacts between the fatty acid substrate and cyclooxygenase channel residues compensate for the loss of ionic interactions between the carboxylate moiety and Arg-120 (17). Contrary to the role of Arg-120 in COX-1, our functional studies highlight that no one individual amino acid within the hydrophobic groove is solely responsible for driving high affinity substrate binding to COX-2. Although mutations to Phe-205, Phe-209, Ile-377, and Gly-533 have little influence on the Km for AA, we demonstrate that the side chains lining the hydrophobic groove of COX-2 work collectively to efficiently position fatty acid substrates for subsequent oxygenation.
Analysis of the crystal structure of muCOX-2 in complex with AA indicates that Phe-381 participates in an intricate π-π stacking network with the aromatic side chains of Phe-205, Phe-209, Phe-210, Tyr-385, Phe-470, and Phe-529 (15). Considering the stabilizing nature of π stacking, it is not surprising that substitutions of this aromatic side chain within the hydrophobic groove rendered the mutant constructs of Phe-381 unstructured and nonfunctional (34). Similarly, the Cγ2 atom of Ile-377 makes a single hydrophobic contact with the side chain of Phe-210, while the Cγ1 and Cδ1 atoms make three hydrophobic contacts with the side chain of Phe-209. Substitutions of Ile-377 with alanine or phenylalanine would disrupt these contacts and serve to destabilize the π-π stacking network. In agreement with our functional results, the conservative mutations I377V and I377L maintain these important structural interactions. Thus, Phe-381 and Ile-377 are critical in maintaining the π-π stacking network, the structural integrity of the hydrophobic groove, and in turn the position of the side chain of Tyr-385 in its catalytically poised conformation.
Previous studies have shown that substitutions at Gly-533 result in significant decreases in the oxygenation of AA by COX-1 and COX-2 (9, 19, 20, 32, 35). The structural consequences for the observed decrease in activity by these mutants was suggested to be a result of the addition of steric bulk to the hydrophobic groove that prevented complete insertion of the ω-end, resulting in misalignment of the pro-S hydrogen of carbon 13 below Tyr-385. However, it was never established whether or not AA could even bind to Gly-533 mutant constructs. The G533V·AA crystal structure presented here illustrates that AA does bind to this construct, and the resulting C-shaped conformation provides a clear explanation of why G533V muCOX-2 is unable to oxygenate AA. AA binds in a novel nonproductive conformation to G533V muCOX-2 such that carbon 13 is misaligned below Tyr-385. It is interesting to note that the observed C-shaped conformation represents another nonproductive pose for AA, one that differs from the nonproductive orientation of AA observed in monomer A of the muCOX-2·AA crystal structure (15). This observation further emphasizes that AA and other fatty acid substrates can bind in a multitude of productive and nonproductive conformations within the cyclooxygenase channel of COX-2.
Despite oxygenating AA at ∼8% the rate of wild type enzyme, G533A muCOX-2 still generates normal PG products (20). Further analysis of the G533V·AA crystal structure provides a plausible explanation for the observed activity of the G533A mutant construct. Specifically, the side chain of Leu-531 exhibits an altered rotamer conformation in the crystal structure compared with the position of this side chain when AA is bound productively in the cyclooxygenase channel (15). The observed rotamer conformation for Leu-531 in the G533V·AA crystal structure is also identical to that seen in the crystal structure of 1-arachidonoyl glycerol bound to muCOX-2 (14) and serves the same purpose, to expand the volume of the cyclooxygenase channel at its opening to accommodate the carboxylate end of AA. As such, we envision that AA could bind within the cyclooxygenase channel of a G533A mutant construct with its ω-end penetrating deeply enough into the hydrophobic groove to position carbon 13 below Tyr-385. Although the alanine substitution would not allow full penetration into the hydrophobic groove, the carboxylate end of AA could be accommodated at the channel opening through the movement of the side chain of Leu-531, resulting in the formation of PGG2. Considering that flexibility associated with the side chain of Leu-531 has not been documented in COX-1, this phenomenon could also explain why a G533A mutant construct in COX-1 does not retain cyclooxygenase activity (19).
Functional analyses and structural characterizations have shown that the side chain of Arg-120 has divergent roles in COX-1 and COX-2. Substitution of Arg-120 with glutamine in ovCOX-1 results in the loss of an ionic interaction between Arg-120 and the carboxylate of AA, leading to a 1000-fold increase in Km (11, 18). Moreover, no PG products were formed from an R120Q COX-1 mutant when αLA was utilized as a substrate, indicating that the side chain of Arg-120 is an important binding determinant for other fatty acid substrates besides AA (18). Conversely, the carboxylate of AA does not form an ionic interaction with the side chain of Arg-120 when bound within the cyclooxygenase channel of COX-2 as an R120Q mutation has little effect on Km (15, 17). The ability of an R120A muCOX-2 construct to efficiently oxygenate AA with only a 3.4-fold increase in Km in the current study further supports this notion. In addition, the fact that LA, SA, and αLA compete with AA for binding to an R120A mutant construct confirms that these alternate substrates also do not require the side chain of Arg-120 for high affinity binding to COX-2. Hence, fatty acid substrate binding to COX-2 appears to be driven via coordinated interactions between substrate and all of the residues lining the channel rather than by a single determinant. We further speculate that the observed decrease in the rates of oxygenation of fatty acid substrates by the R120A muCOX-2 mutant is due to the involvement of the side chain of Arg-120 in the proper alignment of the bisallylic carbon below Tyr-385 for initiation of catalysis.
The oxygenation of αLA by COX-2 results in the production of 12-hydroxy-(9Z,13E/Z,15Z)-octadecatrienoic acid (supplemental Fig. S2) (7). Laneuville et al. (7) speculated that the formation of 12-hydroxy-(9Z,13E/Z,15Z)-octadecatrienoic acid by COX-2 was initiated by hydrogen abstraction from the ω-5 bisallylic carbon of αLA. They reasoned that the carbon chain of the substrate would be too short to align the ω-8 bisallylic carbon below Tyr-385 given that the carboxylate of αLA would be required to interact with the side chain of Arg-120 at the opening of the channel. The structure of αLA bound to muCOX-2 presented here identifies the ω-8 bisallylic carbon as being optimally positioned below Tyr-385 rather than the ω-5 bisallylic carbon. The fact that the carboxylate of αLA does not form an ionic interaction with the side chain of Arg-120 provides the substrate more conformational freedom within the cyclooxygenase channel to fully insert the ω-end into the hydrophobic groove. We speculate that LA and SA bind within the cyclooxygenase channel of COX-2 in a similar fashion.
The crystal structure of αLA bound to muCOX-2 represents the first example of an 18-carbon fatty acid substrate bound within the cyclooxygenase channel of COX-2. As is the case with COX-1, the carbon length of fatty acid substrate does not govern how deeply the ω-end gets inserted into the hydrophobic groove in its productive conformation (36). Although αLA is two carbons shorter than AA, carbon 18 abuts Ile-377 in a manner similar to that observed for carbon 20 of AA, EPA, and 1-arachidonoyl glycerol and carbon 22 of docosahexaenoic acid (14, 15). We speculate that Ile-377 acts as a bumper that assists in the alignment of the proper bisallylic carbon below Tyr-385 for hydrogen abstraction. The only known fatty acid that does not fully penetrate the hydrophobic groove is PA (4). PA, which has 16 carbons and no double bonds, is not a substrate for COX-1 or COX-2 and thus would not require positioning of a bisallylic carbon for cyclooxygenase catalysis.
LA, αLA, and SA compete with AA for binding to the cyclooxygenase channel in both wild type and R120A muCOX-2 constructs, resulting in the inhibition of AA oxygenation. The inhibition of AA oxygenation by these 18-carbon fatty acid substrates was more pronounced in the R120A mutant. As the Km value for AA is increased 3.4-fold for the R120A construct, we speculate that the substrate binding affinities of LA, αLA, and SA become more competitive with AA when compared with wild type enzyme. We also demonstrate that PA can potentiate the rate of oxygenation of these 18-carbon fatty acid substrates in addition to AA. Consistent with our previous structural studies, activation is dependent on the interaction of PA with the side chain of Arg-120 as the R120A mutant construct is unable to potentiate the oxygenation of AA (4).
The observed competition between AA and the 18-carbon fatty acids utilized in this study for binding to COX-2 suggests that ingestion of high levels of essential fatty acids could decrease the production of AA-derived proinflammatory PGs in vivo. Novak and Innis (37) recently showed that healthy adult men and women on unrestricted normal diets consume on average ∼1.7-fold more EPA and docosahexaenoic acid and ∼100-fold more LA and αLA compared with AA on a daily basis. Given these high essential fatty acid:AA ratios, it is intriguing to speculate that essential fatty acids could vie with AA for binding to COX-2 to reduce the synthesis of proinflammatory PGs. This may potentially explain one of the many beneficial health effects of LA and αLA supplementation (38).
X-ray diffraction experiments were conducted at the Cornell High Energy Synchrotron Source (CHESS), supported by National Science Foundation Award DMR-0225180, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, supported by National Institutes of Health Award RR-01646.
↵1 Supported by graduate fellowships from KeyBank.
↵* This work was supported, in whole or in part, by National Institutes of Health Grant GM077176.
↵ This article contains supplemental Tables S1–S4 and Figs. S1 and S2.
This article contains supplemental Tables S1–S4 and Figs. S1 and S2.
The atomic coordinates and structure factors (codes 3TZI and 4E1G) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
↵3 The abbreviations used are: