| Synthesis and degradation of proteoglycans
|
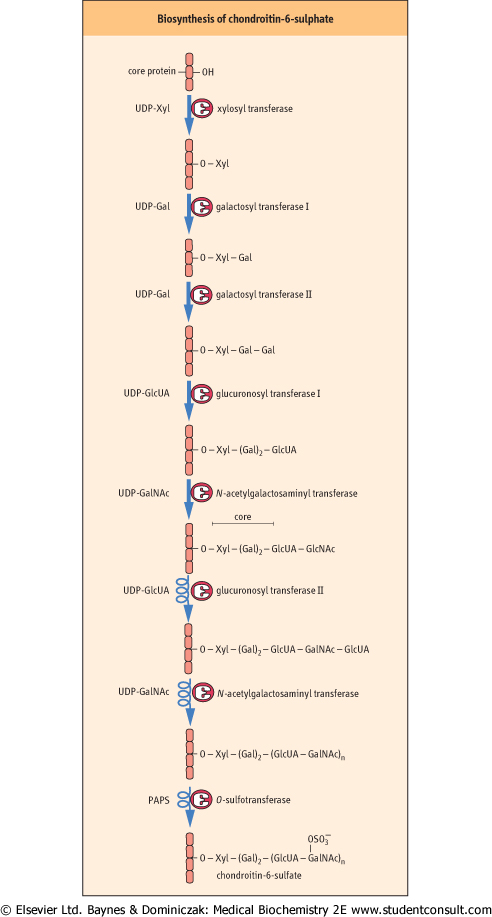
| Proteoglycans are synthesized by a series of glycosyl transferases, epimerases and sulfotransferases, beginning with the synthesis of the core oligosaccharide while the core protein is still in the RER. Synthesis of the repeating oligosaccharide and other modifications takes place in the Golgi apparatus. As with the synthesis of glycoproteins and glycolipids, separate enzymes are involved in individual steps. For example, there are separate galactosyl transferases for each of the galactose units in the core, a separate GlcUA transferase for the core and repeating disaccharides, and separate sulfotransferases for the C-4 and C-6 positions of the GalNAc residues of chondroitin sulfates. Phosphoadenosine phosphosulfate (PAPS) (see Fig. 26.6) is the sulfate donor for the sulfotransferases. These pathways are illustrated in Figure 27.7, for chondroitin-6-sulfate.
|
| Defects of proteoglycan degradation lead to mucopolysaccharidoses
|
Figure 27.7 Synthesis of the proteoglycan, chondroitin-6-sulfate. Several enzymes participate in this pathway. Xyl, xylose . . |
| page 395 |  | | page 396 |
| MECHANISM OF ANTICOAGULATION EFFECT OF HEPARIN |
| Heparin is heterogeneous (3,000-30,000 kDa), polyanionic oligosaccharide activator of antithrombin III (AT). AT is a slow, but quantitatively important inhibitor of thrombin (Factor X) and other factors (IX, XI, XII) in the blood-clotting cascade. When heparin binds to AT, it converts AT from a slow inhibitor to a rapid inhibitor of coagulating enzymes. Heparin interacts with a lysine residue in AT and induces a conformational change that promotes covalent binding of AT to the active serine centers of coagulating enzymes, inhibiting their pro-coagulant activity. Heparin then dissociates from the ternary complex and can be recycled for anticoagulation. The smallest, most active component of heparin is a pentasaccharide that has a Kd of ∼10 μmol/L): GlcN-(N-sulfate-6-O-sulfate)-α1,4-GlcUA-β1,4-GlcN-(N-sulfate-3,6-di-O-sulfate)-α1,4-IdUA-(2-O-sulfate)-α-1,4-GlcN-(N-sulfate-6-O-sulfate-). Heparin has an average half-life of 30 min in the circulation, so that it is commonly administered by infusion. Heparin does not have fibrinolytic activity; therefore, it will not lyse existing clots. In addition to its anticoagulant activity, heparin also releases several enzymes from proteoglycan binding sites on the vascular wall, including lipoprotein lipase, which is often assayed as heparin-releasable plasma lipoprotein lipase activity. Lipoprotein lipase is inducible by insulin, and decreased activity of this enzyme delays plasma clearance of chylomicrons and VLDL, contributing to hypertriglyceridemia in diabetes. |
| Figure 27.8 Degradation of heparan sulfate. This proceeds by a defined sequence of lysosomal hydrolase activities. |
|
Table 27-3.
Enzymatic defects characteristic of various mucopolysaccharidoses. |
| Body_ID: None |
| The mucopolysaccharidoses |
| Body_ID: T027003.50 |
| Syndrome | Deficient enzyme | Product accumulated in lysosomes and secreted in urine |
| Body_ID: T027003.100 |
| Hunter's | iduronate sulfatase | heparan and dermatan sulfate |
| Body_ID: T027003.150 |
| Hurler's | α-iduronidase | heparan and dermatan sulfate |
| Body_ID: T027003.200 |
| Morquio's A | galactose-6-sulfatase | keratan sulfate |
| Body_ID: T027003.250 |
| B | β-galactosidase | keratan sulfate |
| Body_ID: T027003.300 |
| Sanfilippo's A | heparan sulfamidase | heparan sulfate |
| Body_ID: T027003.350 |
| B | N-acetylglucosaminidase | |
| Body_ID: T027003.400 |
| C | N-acetylglucosamine-6-sulfatase | |
| Body_ID: T027003.450 |
| The degradation of proteoglycans occurs in lysosomes. The protein portion is degraded by lysosomal proteases, and the GAG chains are degraded by the sequential action of a
number of different lysosomal acid hydrolases. The stepwise degradation of GAGs involves exoglycosidases and sulfatases, beginning from the external end of the glycan chain. This may involve the removal of sulfate by a sulfatase, then removal of the terminal sugar by a specific glycosidase, and so on. Figure 27.8 shows the steps in the degradation of
dermatan sulfate. As with degradation of glycosphingolipids, if one of the enzymes involved in the stepwise pathway is missing, the entire degradation process is halted at that point, and the undegraded molecules accumulate in the lysosome. The lysosomal storage diseases resulting from accumulation of GAGs are known as mucopolysaccharidoses (Table 27.3), because of the original designation of GAGs as mucopolysaccharides. There are more than a dozen such mucopolysaccharidoses, resulting from defects in degradation of GAGs. In general, these diseases can be diagnosed by the identification of specific GAG chains in the urine, followed by assay of the specific hydrolases in leukocytes or fibroblasts. Although they are rare genetic diseases, the mucopolysaccharidoses have been important in the elucidation of the role of lysosomes in health and disease, and the mechanisms involved in biosynthesis of lysosomes and targeting of lysosomal enzymes.
|
|