| Low substrate specificity of hepatic enzymes produces a wide-ranging capability for drug metabolism
|
| Most drugs are metabolized by the liver. Among other effects, this hepatic metabolism usually increases the hydrophilicity of drugs, and therefore their ability to be excreted. Generally, the metabolites that are produced are less pharmacologically active than the substrate drug; however, some inactive pro-drugs are converted to their active forms as a result of processing in the liver. It is necessary for the hepatic drug-metabolizing systems to act on an infinite range of molecules; this is achieved by the enzymes involved having low substrate specificity. Metabolism proceeds in two phases:
|
- phase I: the polarity of the drug is increased by oxidation or hydroxylation catalyzed by a family of microsomal cytochrome P450 oxidases;
- phase II: cytoplasmic enzymes conjugate the functional groups introduced in the first phase reactions, most often by glucuronidation or sulfation.
|
| Three of the 12 cytochrome P450 gene families share the responsibility for drug metabolism
|
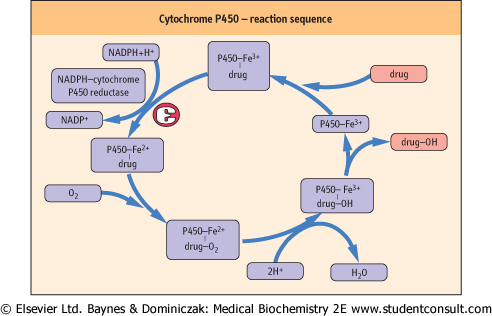
| The cytochrome P450 enzymes are heme-containing proteins that co-localize with reduced nicotinamide adenine dinucleotide phosphate (NADPH) cytochrome P450 reductase. The reaction sequence catalyzed by these enzymes is shown in Figure 28.6. There are 12 cytochrome P450 gene families, of which three, designated CYP1, CYP2, and CYP3, are mainly responsible for drug metabolism. Each gene locus has multiple alleles, and the P450 enzyme encoded by the CYP3A4 gene appears to be involved in the metabolism of most drugs. Liver disease is likely to impair drug metabolism, and so drugs must be prescribed carefully for such patients.
|
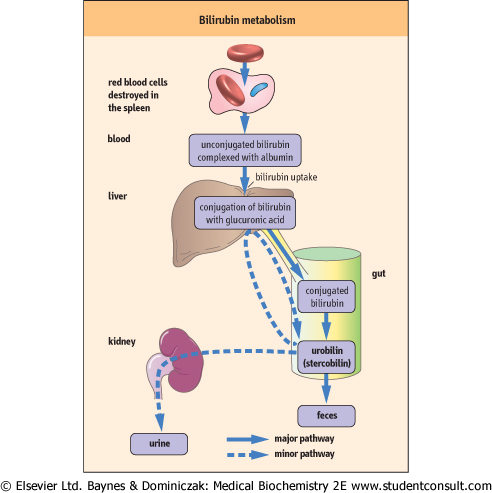
| Figure 28.5 Normal bilirubin metabolism. |
| page 405 |  | | page 406 |
| Figure 28.6 The role of cytochrome P450 system in the metabolism of drugs. |
| The hepatic synthesis of P450 cytochromes is increased by certain drugs and other xenobiotic agents - a process known as 'induction', which increases the rate of phase I reactions. Conversely, drugs that form a relatively stable complex with a particular cytochrome P450 inhibit the metabolism of other drugs that are normally substrates for that cytochrome P450. Allelic variation that affects the catalytic activity of a cytochrome P450 will also affect the pharmacologic activity of drugs. The best described example of such polymorphism is that of the P450 cytochrome CYP2D6, which was recognized initially in the 5-10% of normal individuals who were noted to be slow to hydroxylate debrisoquine, a now little-used blood-pressure-lowering drug. CYP2D6 also metabolizes a significant number of other commonly used drugs, so that 'debrisoquine polymorphism' remains clinically significant.
|
|