| ERYTHROPOIETIN TREATMENT FOR ANEMIA |
| Anemia, defined clinically as a decrease in hematocrit (<37%) or Hb concentration (<120 g/L), may be caused by blood loss, excessive hemolysis, and/or deficient erythropoiesis. One therapeutic strategy in the treatment of some anemias is the production of new erythrocytes, a bone marrow stem cell proliferation and differentiation process that requires the hormone erythropoietin (EPO). In the 1980s and 1990s clinical trials of recombinant human EPO (rHuEPO, Epogen®, Procrit®) showed that exogenous hormone was particularly effective in increasing Hb levels in patients with anemias associated with chronic renal failure, chemotherapy for nonmyeloid malignancies, and treatment of HIV infections. |
| EPO is secreted by renal peritubular endothelial cells in response to localized tissue hypoxia. Upon binding to receptors that belong to the cytokine superfamily, EPO initiates multiple signaling pathways in erythroid progenitor cells. The glycoprotein EPO contains both N-linked and O-linked oligosaccharides. Because the oligosaccharide groups exhibit microheterogeneity in sugar content and structure, particularly in the number of N-acetylneuraminic (sialic) acid residues, a family of isoforms is always observed by isoelectric focusing polyacrylamide electrophoresis. Additional heterogeneity is seen between natural EPO and rHuEPO that is produced commercially in Chinese hamster ovary cells. Darbopoetin alfa (Aranesp®), a second-generation rHuEPO, has been 'glycoengineered' to contain additional N-linked oligosaccharides, structural features that significantly enhance its stability and bioactivity. |
| Sickle cell disease is caused by an inherited structural abnormality in the β-globin polypeptide. Clinically, an
individual with sickle cell disease presents with intermittent episodes of hemolytic and painful vaso-occlusive crises, the latter leading to severe pain in bones, chest, and abdomen. Common side effects include impaired growth, increased susceptibility to infections, and multiple organ damage. Sickle cell disease has a prevalence of 40% in some regions of equatorial Africa; among black Americans, heterozygotic carriers number 8%, yielding about 0.2% frequency of the disease in the African American population. The Hb molecule in sickle cell disease (HbS) has been studied biochemically and biophysically for over 50 years, and sickle cell disease has become the paradigm of a molecular disease.
|
| page 45 |  | | page 46 |
|
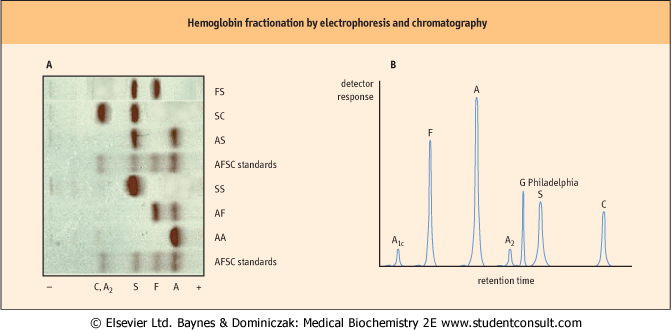
| Figure 4.8 Normal and abnormal hemoglobins can be separated by electrophoretic and chromatographic methods. (A) This panel shows cellulose acetate electrophoresis (pH 8.4) of blood samples obtained for neonatal screening. This rapid technique will tentatively identify HbS and HbC, two common mutant hemoglobins in the African American population. Additional tests are required for a definitive diagnosis. FS, newborn with sickle cell disease; SC, double heterozygote child with sickle cell-like disease; AS, child with sickle cell trait; AF, normal neonate. (B) This trace illustrates high-pressure liquid chromatography (HPLC) with a cation-exchanger solid phase, a technique capable of separating and quantifying more than 40 hemoglobins. HPLC may also be used to measure HbA1c, a glycated protein that provides information on the progression and treatment of diabetes mellitus (Chapter 20). Also shown is the elution profile of Hb G Philadelphia (Asn68α → Lys), a common but benign variant that co-migrates with HbS on electrophoresis. |
| SEPARATION OF HEMOGLOBIN VARIANTS AND MUTANTS AND THE DIAGNOSIS OF HEMOGLOBINOPATHIES |
| The mobility of a protein during electrophoresis or chromatography is determined by its charge and interaction with the matrix. Three commonly used techniques provide sufficient resolution to separate hemoglobin variants differing in a single charge from HbA: electrophoresis, isoelectric focusing, and ion exchange chromatography. Electrophoretic and chromatographic separations of Hb are illustrated in Figure 4.8. The volume of hemolysate required (<100 μL) makes these techniques suitable for neonate and adult blood samples. Quantification is performed by scanning densitometry or absorption spectrometry. Indications of abnormalities in screening tests are followed by complete blood count (see Table 4.2), additional protein analysis, and DNA analysis to identify specific mutations to the globin genes. |
| ANALGESIC TREATMENT OF SICKLE CELL VASO-OCCLUSIVE CRISES |
Vaso-occlusive pain is the most common problem reported by individuals with sickle cell disease. It is also the most frequent reason for emergency-room treatment and hospital admission of individuals with sickle cell disease. Episodes of vaso-occlusive pain are unpredictable and are often excruciating and incapacitating. The origin of this progressive pain involves altered rheologic and hematologic properties of erythrocytes attributable to HbS polymerization and aggregation, coupled with an inflammatory leukocytosis and elevation of plasma acute-phase proteins (Chapter 3). The pain lasts an average of 4-10 days. To provide relief to the patient, nonnarcotic, narcotic, and adjuvant analgesics are used alone or in combination. Obviously, the severity and duration of the pain dictate the most appropriate analgesic regimen. Several recent studies suggest additional options for the patient and physician: continuous intravenous infusion of a nonsteroidal anti-inflammatory drug (ketoralac) reduced the requirement for opioid analgesic (meperidine); and continuous epidural administration of local anesthetic (lidocaine ) and opioid analgesic (fentanyl) effectively decreased pain that was unresponsive to conventional measures. ) and opioid analgesic (fentanyl) effectively decreased pain that was unresponsive to conventional measures. |
| page 46 | | | page 47 |
| A PATIENT WITH MUTANT HEMOGLOBIN |
| During a complete physical examination related to diabetes mellitus and cataracts, a 66-year-old male presented with cyanosis of the lips. The patient was found to have mild hypertension, left ventricular hypertrophy, and other electrocardiographic abnormalities. Since childhood, he has followed a restricted exercise program because of suspected heart disease. Laboratory findings indicated increases in hematocrit (HCT), erythrocyte count, and Hb concentration (see Table 4.2). Platelet and leucocyte counts were normal, as was pulmonary function. The P50 of the patient's whole blood was 58 mmHg (normal = 27 ± 2); the Hill coefficient was 1.6 (normal = 2.7 ± 0.2). The possibility of an abnormal Hb was considered. Biochemical analysis of his Hb revealed a mutant β-globin polypeptide in which Asn102 β was replaced by Thr. Electrophoretic analysis indicated that the patient was heterozygous for Hb Kansas. |
| Comment. Not only does this mutant Hb exhibit a dramatically reduced affinity for O2 (increased P50), but subunit cooperativity is also markedly disrupted (decreased n). Examination of the tertiary structures of deoxygenated and oxygenated Hb provides an explanation: Asp94α hydrogen bonds to Asn102β in the heterodimer interface, a bond that only exists in the R-state (Fig. 4.5). These residues are too far apart in the T-state to create a noncovalent interaction. In Hb Kansas the absence of this critical oxygenated Hb-stabilizing bond shifts the equilibrium between the T- and R-states toward deoxygenation, less communication among the four ligand-binding sites, and a decrease in subunit cooperativity. Although Hb Kansas is very rarely seen in a clinical setting, the phenotypes of this and other rare hemoglobinopathies continue to provide valuable insight into protein structure-function relationships. |
|
| HbA remains a true solute at rather high concentrations, largely as a result of a polar exterior surface that is compatible with and nonreactive with nearby Hb molecules. In contrast, HbS, when deoxygenated, is less soluble. It forms long, filamentous polymers that readily precipitate, distorting erythrocyte morphology to the characteristic sickle shape. The mutation is Glu6 β→Val: a surface-localized charged amino
acid is replaced by a hydrophobic residue. Valine on the mutant β-globin subunit fits into a complementary pocket (sometimes called a 'sticky patch') formed on the β-globin subunit of another Hb molecule, a pocket that becomes exposed only upon the release of bound O2 in tissue capillaries. In the homozygous individual with sickle cell disease (HbS/HbS), the complex process of nucleation and polymerization occurs rapidly, producing about 10% of circulating erythrocytes that are sickled. In the heterozygous individual (HbA/HbS, sickle cell trait), the kinetics of sickling are decreased by at least a factor of 103, thereby accounting for the asymptomatic nature of this genotype. In dilute solution, HbS has interactions with O2 (P50 value, Hill coefficient) that are similar to those for HbA. However, the Bohr effect on concentrated HbS is more pronounced, leading to greater release of O2 in the capillaries and increased propensity for polymer growth.
|
| Sickled erythrocytes exhibit less deformability; they no longer move freely through the microvasculature and often block blood flow, especially in the spleen and joints. Moreover, these cells lose water, become fragile, and have a considerably shorter life span, leading to hemolysis and anemia. Except during extreme physical exertion, the heterozygous individual appears normal. For reasons that remain to be elucidated, heterozygosity is associated with an increased resistance to malaria, specifically growth of the infectious agent Plasmodium falciparum in the erythrocyte. This observation represents an example of a selective advantage that the HbA/HbS heterozygote exhibits over either the HbA/HbA normal or the HbS/HbS homozygote and probably explains the persistence of HbS in the gene pool.
|
|