| Mobilization of lipids during gluconeogenesis and work
|
| page 195 |  | | page 196 |
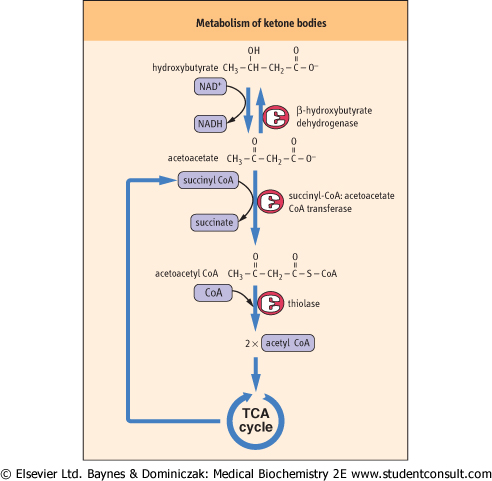
| Figure 14.8 Catabolism of ketone bodies in peripheral tissues. Succinyl-CoA: acetoacetate CoA transferase activates the transfer of acetoacetate to acetoacetyl CoA. A thiokinase-type enzyme may also directly activate acetoacetate in some tissues. |
|
Table 14-3.
Plasma concentrations of fatty acids and ketone bodies in different nutritional states. |
| Body_ID: None |
| Plasma concentrations of fatty acids and ketone bodies |
| Body_ID: T014003.50 |
| Substrate | Plasma concentration (mmol/L) | |
| Body_ID: T014003.100 |
| | Normal | Fasting | Starvation |
| Body_ID: T014003.150 |
| Fatty acids | 0.6 | 1.0 | 1.5 |
| Body_ID: T014003.200 |
| Acetoacetate | >0.1 | 0.2 | 1-2 |
| Body_ID: T014003.250 |
| β-Hydroxybutyrate | >0.1 | 1 | 5-10 |
| Body_ID: T014003.300 |
| HELLP AND AFLP SYNDROMES IN MOTHERS OF CHILDREN BORN WITH LCHAD (INCIDENCE 1 IN 200,000) |
| Long chain L-3-hydroxy-acyl-CoA dehydrogenase deficiency (LCHAD) can present in a wide variety of ways. Those affected are prone to episodes of non-ketotic hypoglycaemia, but may develop fulminant hepatic failure, cardiomyopathy, rhabdomyolysis, and occasionally neuropathy and retinopathy. As with deficiencies in MCAD or LCAD, treatment involves avoidance of fasting and diets enriched in medium chain fatty acids. |
| Perhaps the most striking feature of this rare defect in fatty acid metabolism is the association with maternal HELLP (haemolysis, elevated liver enzymes and low platelets) and AFLP (acute fatty liver of pregnancy). These potentially fatal obstetric emergencies may occur in mothers who are heterozygotes for LCHAD, especially if the child has LCHAD. These syndromes are also associated with another recessive fatty acid defect, carnitine palmitoyl-transferase-I deficiency. The pathophysiological mechanisms have not been elucidated. |
Insulin, glucagon, epinephrine , and cortisol control the direction and rate of glycogen and glucose metabolism in liver. During fasting and starvation, hepatic gluconeogenesis is
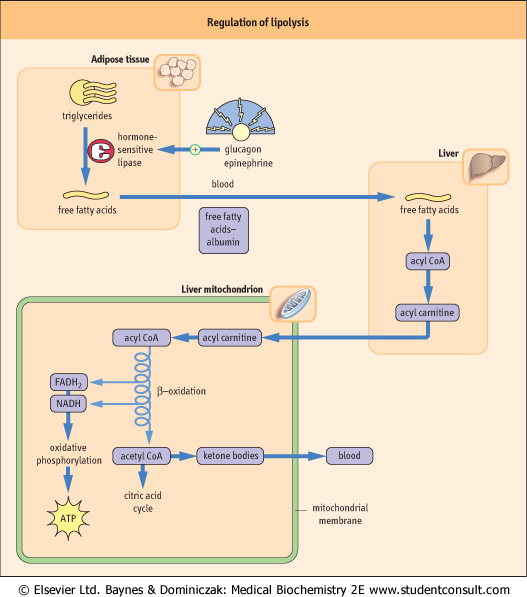
activated by glucagon and requires the coordinated degradation of proteins and release of amino acids from muscle, and the degradation of triglycerides and release of fatty acids from adipose tissue. This process, known as lipolysis, is controlled by the adipocyte enzyme hormone-sensitive lipase, which is activated by phosphorylation by cAMP-dependent protein kinase A in response to increasing plasma concentrations of glucagon (see Chapter 20). Like gluconeogenesis, lipolysis is inhibited by insulin. , and cortisol control the direction and rate of glycogen and glucose metabolism in liver. During fasting and starvation, hepatic gluconeogenesis is
activated by glucagon and requires the coordinated degradation of proteins and release of amino acids from muscle, and the degradation of triglycerides and release of fatty acids from adipose tissue. This process, known as lipolysis, is controlled by the adipocyte enzyme hormone-sensitive lipase, which is activated by phosphorylation by cAMP-dependent protein kinase A in response to increasing plasma concentrations of glucagon (see Chapter 20). Like gluconeogenesis, lipolysis is inhibited by insulin.
|
| The activation of hormone-sensitive lipase has predictable effects - increasing the concentration of free fatty acids, glycerol, and ketone bodies in plasma during fasting and starvation (Fig. 14.9); similar effects are observed in response to epinephrine during the stress response. Epinephrine activates both glycogenolysis in the liver and lipolysis in adipose tissue, so that both fuels, glucose and fatty acids, increase in blood during stress. Cortisol exerts a more chronic effect on lipolysis and also causes insulin resistance. Cushing's syndrome (see Chapter 37), in which there are high blood concentrations of cortisol, is characterized by hyperglycemia, muscle wastage, and redistribution of fat from glucagon-sensitive adipose depots to atypical sites, such as the cheeks, upper back, and trunk.
|
| page 196 | | | page 197 |
| Figure 14.9 Regulation of lipid metabolism by glucagon and epinephrine. Glucagon and epinephrine activate hormone-sensitive lipase in adipose tissue, in coordination with activation of proteolysis in muscle and gluconeogenesis in liver. Metabolism of fatty acids through β-oxidation in liver yields ATP for gluconeogenesis. The acetyl-CoA is converted to and released to blood as ketone bodies. These effects are reversed by insulin following a meal. |
|