| Proteins that transport metal ions
|
| The ability of proteins to bind and transport metal ions is of major importance
|
Iron is an essential element for many metabolic processes, and is an important component of the heme proteins, myoglobin, hemoglobin, and cytochromes. Within the plasma, iron is transported bound to transferrin as ferric ions (Fe3+) and is released from the protein into tissues after it has bound to specific cell receptors and the resulting complex has been internalized. The iron is then deposited in storage sites as ferritin or hemosiderin, or is used in synthesis of heme proteins. The binding of ferric ions to transferrin protects against the toxic effects of these ions. In inflammatory reactions, the iron-transferrin complex is degraded by the reticuloendothelial system without a corresponding increase in the synthesis of either of its components; this results in low plasma concentrations of transferrin and iron.
- Ferritin is the major iron storage protein found in almost all cells of the body. It acts as the reserve of iron in the liver and bone marrow. The concentration of ferritin in plasma is proportional to the amount of stored iron, and so measurement of plasma ferritin is one of the best indicators of iron deficiency;
- Hemosiderin is a derivative of ferritin and is found in the liver, spleen, and bone marrow. It is insoluble in aqueous solutions, and forms aggregates that slowly release iron when deficiency exists;
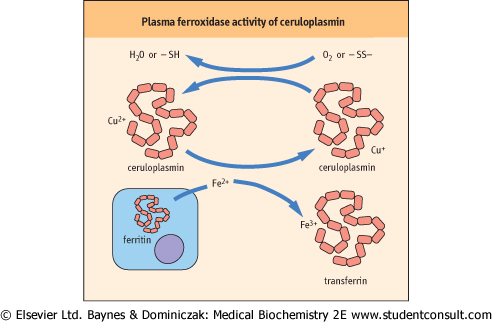
- Ceruloplasmin is the major transport protein for copper, an essential trace element. Ceruloplasmin helps export copper from the liver to peripheral tissues, and is essential for the regulation of the oxidation-reduction reactions, transport, and utilization of iron (Fig. 3.5). Increased concentrations of ceruloplasmin occur in active liver disease and in tissue damage.
|
Low concentrations of ceruloplasmin and total copper are observed in Wilson's disease, the autosomal recessive disorder of copper transport. The genetic defect is on chromosome 13 resulting in a mutation in copper-transporting P-type adenosine triphosphatase (ATP7B) which is responsible for transport of copper into ceruloplasmin and elimination of copper through bile. Mutation of ATP7B results in the accumulation of copper in the liver, brain and kidney. These mutations are readily identified by haplotype and mutational analysis and so genetic screening of patients and their families for Wilson's disease is possible. triphosphatase (ATP7B) which is responsible for transport of copper into ceruloplasmin and elimination of copper through bile. Mutation of ATP7B results in the accumulation of copper in the liver, brain and kidney. These mutations are readily identified by haplotype and mutational analysis and so genetic screening of patients and their families for Wilson's disease is possible.
|
| A 14-year-old girl was admitted as an emergency. She was jaundiced with abdominal pain and had an enlarged, tender liver with drowsiness and asterixis (flapping tremor) due to acute liver failure. Previous history revealed behavior disturbance, difficulty with movement in the recent past, and truancy from school. Her ceruloplasmin concentration was 50 mmol/L (normal range 200-450 mmol/L [20-45 mg/dL]), serum copper was 8 mmol/L (normal range 13-19 μmol/L [80-120 μg/dL]), urinary excretion of copper was 2.2 mmol/ 24 h (normal range 2-3.9 μmol/24 h [13-25 μg/dL]), and a liver biopsy established the diagnosis of Wilson's disease. |
| Comment. This case highlights the importance of measurement of ceruloplasmin. In Wilson's disease, a deficiency of ceruloplasmin results in low plasma concentrations of copper. The metabolic defect is in the excretion of copper in bile and its reabsorption in the kidney; copper is deposited in liver, brain, and kidney. Liver symptoms are present in patients of younger age, and cirrhosis and neuropsychiatric problems are manifest in those who are older. Detection of low plasma concentrations of ceruloplasmin and copper, increased urinary excretion of copper, and markedly increased concentrations of copper in the liver confirms the diagnosis. |
| page 29 |  | | page 30 |
| Figure 3.5 Plasma ferroxidase activity of ceruloplasmin. Oxidation of Fe2+ by ceruloplasmin permits the binding and transport of iron by plasma transferrin. The cuprous ion (Cu2+) bound to ceruloplasmin is regenerated by reaction with oxygen or with oxidized thiol groups. |
|