| Examples of transport systems and their coupling
|
| Ca2+transport and mobilization in muscle
|
| Striated muscle (skeletal and cardiac) is composed of bundles of multinucleated muscle cells (see Chapter 19). Each cell is packed with bundles of actin and myosin filaments (myofibrils) that produce contraction. During muscle contraction, nerves at the neuromuscular junction stimulate local depolarization of the membrane by opening voltage-dependent Na+ channels. The depolarization spreads rapidly into invaginations of the plasma membrane called the transverse (T) tubules, which extend around the myofibrils (see Fig. 19.5).
|
| page 87 |  | | page 88 |
| ABC TRANSPORTERS AND DISEASES |
| Human genome data suggests that there are at least 48 genes for ABC transporters. An unusually wide range of diseases are caused by defects of ABC transporters, including Tangier disease, Stargardt disease, progressive intrahepatic cholestasis, Dubin-Johnson syndrome, pseudoxanthoma elasticum, familial persistent hyperinsulinemic hypoglycemia of infancy (PHHI), adrenoleukodystrophy, Zellweger syndrome, sitosterolemia and cystic fibrosis. |
| Comment. Cystic fibrosis (CF) is the most common potentially lethal autosomal recessive disease of caucasian populations, affecting 1 in 2500 newborns. CF is usually manifest as exocrine pancreatic insufficiency, an increase in the concentration of chloride ions (Cl-) in sweat, male infertility, and airway disease. The last of these leads to progressive lung dysfunction, which is the major cause of morbidity and mortality. CF is caused by mutations in the gene encoding a Cl- channel named CFTR (cystic fibrosis transmembrane conductance regulator). ATP binding to the CFTR is required for channel opening. The lack of this channel activity in epithelia of CF patients is believed to be central to the pathogenesis of the disease. |
The ATP-sensitive K+ channel (KATP) participates in regulation of insulin secretion in pancreatic islet β-cells. When the blood concentration of glucose increases, glucose is transported into the β-cell through a glucose transporter (GLUT-2) and metabolized, resulting in an increase in cytoplasmic ATP concentration. The ATP binds to the regulatory subunit of the K+ channel, KATP-β (called the sulfonylurea receptor, SUR1) causing structural change of a KATP-α subunit, which closes the KATP channel. This induces depolarization of the plasma membrane (decreased voltage gradient across the membrane) and activates voltage-dependent calcium (Ca2+) channels (VDCCs). The entry of Ca2+ stimulates exocytosis of vesicles that contain insulin. The binding of sulfonylureas such as tolubutamide and glibenclamide to KATP-β on the outside of the plasma membrane is thought to mimic the regulatory effect of intracellular ATP. Sulfonylureas stimulate insulin secretion, which decreases blood glucose concentration in diabetes. Defective KATP channels, which are unable to transport K+, induce low blood glucose concentration - a condition called PHHI that occurs in 1 per 50000 persons - as a result of loss of K+-channel function and continuous insulin secretion. increases, glucose is transported into the β-cell through a glucose transporter (GLUT-2) and metabolized, resulting in an increase in cytoplasmic ATP concentration. The ATP binds to the regulatory subunit of the K+ channel, KATP-β (called the sulfonylurea receptor, SUR1) causing structural change of a KATP-α subunit, which closes the KATP channel. This induces depolarization of the plasma membrane (decreased voltage gradient across the membrane) and activates voltage-dependent calcium (Ca2+) channels (VDCCs). The entry of Ca2+ stimulates exocytosis of vesicles that contain insulin. The binding of sulfonylureas such as tolubutamide and glibenclamide to KATP-β on the outside of the plasma membrane is thought to mimic the regulatory effect of intracellular ATP. Sulfonylureas stimulate insulin secretion, which decreases blood glucose concentration in diabetes. Defective KATP channels, which are unable to transport K+, induce low blood glucose concentration - a condition called PHHI that occurs in 1 per 50000 persons - as a result of loss of K+-channel function and continuous insulin secretion. |
| MEASUREMENT OF GLUCOSE TRANSPORT INTO ERYTHROCYTE |
| Aliquots (0.2 ml) of erythrocyte suspension in phosphate-buffered saline (a hematocrit of 0.60 to 0.80) are mixed with 0.3 ml of the saline containing [14C]3-O-methyl-D-glucose, and incubated at 20°C for short period (2-15 sec). Uptake is interrupted by the rapid addition of 5 ml of ice-cold saline with 10 μM HgCl2 and 100 μM phloretin to inhibit GLUT-1. The mixture is centrifuged at 2000 ×g for 10 min at 4°C. The erythrocyte pellet is washed twice with 5 ml of the same saline, and then lysed in 2.8% trichloroacetic acid (to precipitate protein). Radioactivity is measured in aliquots of the supernatant. The uptake is expressed as picomoles of 3-O-methyl-d-glucose per 106 erythrocyte. Nonspecific uptake is evaluated with radioactive l-glucose. |
| Voltage-dependent Ca2+ channels (VDCC) located in the T tubules of skeletal muscle change their conformation in response to membrane depolarization, and directly activate a Ca2+-release channel in the sarcoplasmic reticulum membrane, a network of flattened tubules that surrounds each myofibril in the muscle-cell cytoplasm. The escape of Ca2+
from the lumen (interior compartment) of the sarcoplasmic reticulum increases the cytoplasmic concentration of Ca2+ (depolarization-induced Ca2+ release) about 100-fold, from 10-4mmol/L (0.0007 mg/L) to about 10-2mmol/L (0.07 mg/dL), triggering ATP hydrolysis by myosin and muscle contraction. A Ca2+-ATPase in the sarcoplasmic reticulum then hydrolyses ATP to transport Ca2+ back out of the cell into the lumen, decreasing the cytoplasmic concentration of Ca2+, and the muscle relaxes (Fig. 7.6, left).
|
| In cardiac muscle, VDCCs permit the entry of a small amount of Ca2+, which induces Ca2+ release through Ca2+ channel from the lumen of the sarcoplasmic reticulum (Ca2+-induced Ca2+ release). Not only the sarcoplasmic reticulum Ca2+-ATPase, but also an Na+/ Ca2+-antiporter and a plasma membrane Ca2+-ATPase are responsible for pumping out cytoplasmic Ca2+ from heart muscle (Fig. 7.6, right). The rapid restoration of ion gradients allows for rhythmic contraction of the heart.
|
| Role of Na+/K+-ATPase in glucose uptake
|
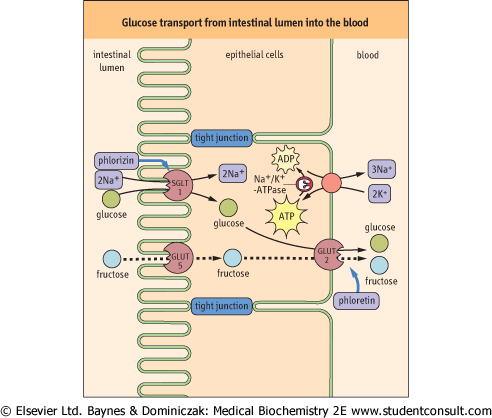
| The transport of blood glucose into cells is generally by facilitated diffusion, as the intracellular concentration of glucose is typically less than that of blood (Table 7.4). In contrast, the transport of glucose from the intestine into blood involves both facilitated diffusion and active transport processes (Fig. 7.7). Active transport is especially important for maximal recovery of sugars from the intestine when the intestinal concentration of glucose falls below that in the blood.
|
| page 88 | | | page 89 |

|
| Figure 7.6 Ca2+ movement in muscle contraction cycle. Roles of transporters in Ca2+ movements in skeletal (left) and cardiac (right) muscle cells during contraction. Thick arrows indicate the binding sites for inhibitors. In skeletal muscle, VDCCs (voltage-dependent calcium channels) directly activate release of Ca2+ from the sarcoplasmic reticulum. The increased cytoplasmic Ca2+ concentration triggers muscle contraction. A Ca2+-ATPase in the sarcoplasmic reticulum transports Ca2+ back into the lumen, decreasing the cytoplasmic Ca2+ concentration, and the muscle relaxes. In heart muscle, VDCCs allow entry of a small amount of Ca2+, which induces release of Ca2+ from the lumen of sarcoplasmic reticulum. Two types of Ca2+-ATPases and an Na+/Ca2+-antiporter are responsible for pumping cytoplasmic Ca2+ out of the muscle cell. The Na+/ Ca2+-antiporter uses the sodium (Na+) gradient produced by Na+/K+-ATPase to antiport Ca2+. DHP = dihydropyridine, nifedipine, a calcium channel blocker used for treatment of hypertension. |
| An Na+-coupled glucose symporter SGLT1, driven by an Na+-gradient formed by Na+/K+-ATPase, transports glucose into the intestinal epithelial cell, while GLUT-2 facilitates the
downhill movement of glucose into the portal circulation (Fig. 7.7).
|
| The kidneys constitute an ultrafiltration system that filters small molecules from blood. However, glucose, amino acids, many ions, and other nutrients in the ultrafiltrate are almost completely reabsorbed in the proximal tubules, by symport processes. Glucose is reabsorbed primarily by sodium glucose transporter 2 (SGLT2) (one-to-one stoichiometry), into renal proximal tubular epithelial cells. Much smaller amounts of glucose are recovered by SGLT1 in a later segment of the tubule, which couples transport of one molecule of glucose to two sodium ions. The concentration of Na+ in the filtrate is 140 mmol/L (322 mg/dL), while that inside the epithelial cells is 30 mmol/L (69 mg/dL), so that Na+ flows 'downhill' along its gradient, dragging glucose 'uphill' against its concentration gradient. As in intestinal epithelial cells, the low intracellular concentration of Na+ is maintained by an Na+/K+-ATPase on the opposite side of the tubular epithelial cell, which antiports three cytoplasmic sodium ions and two extracellular potassium ions, coupled with hydrolysis of a molecule of ATP. Glucose accumulated in the epithelial cell is further transported downhill across the membrane to the blood by GLUT-2 (facilitated diffusion).
|
| Acidification of gastric juice by a proton pump in the stomach
|
| page 89 | | | page 90 |
|
| Figure 7.7 Glucose transport from intestinal lumen into the blood. Glucose is pumped into the cell through the Na+-coupled glucose symporter (SGLT1), and passes out of the cell by facilitated diffusion mediated by the GLUT-2 uniporter. The Na+ gradient for glucose symport is maintained by the Na+/K+-ATPase, which keeps the intracellular concentration of Na+ low. SGLT1 is inhibited by phlorizin and GLUT-2 by phloretin. Phloretin-insensitive GLUT-5 catalyzes the uptake of fructose by facilitated diffusion. The fructose is then exported through GLUT-2. A defect of SGLT1 causes glucose/galactose malabsorption. Adjacent cells are connected by impermeable tight junctions, which prevent solutes from crossing the epithelium. However, leakage of salts (Na+ and Cl-) through tight junctions induces diarrhea as a result of inhibition of water absorption. Diarrhea is also induced by laxatives such as phenolphthalein, which is an irritant cathartic for the colon. Thick arrows indicate the binding sites for inhibitors. |
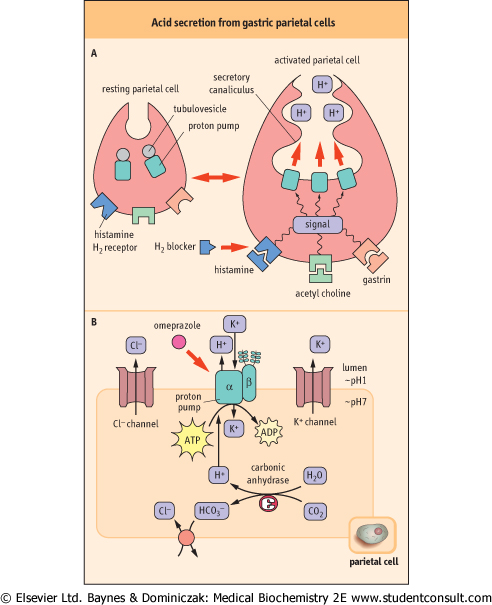
| The lumen of the stomach is highly acidic (pH ∼1) because of the presence of a proton pump (H+/K+-ATPase; P-ATPase
in Table 7.5) that is specifically expressed in gastric parietal cells. The gastric proton pump is localized in intracellular vesicles in the resting state. Stimuli such as histamine and gastrin induce fusion of the vesicles with the plasma membrane (Fig. 7.8A). The pump antiports two cytoplasmic protons and two extracellular potassium ions, coupled with hydrolysis of a molecule of ATP, thus it is called an H+/K+-ATPase. The counter-ion Cl- is secreted through a Cl- channel, producing hydrochloric acid (HCl) (gastric acid) in the lumen (Fig. 7.8B).
|
| Figure 7.8 Acid secretion from gastric parietal cells. (A) Acid secretion is stimulated by extracellular signals and accompanied by morphologic changes in parietal cells [from resting (left) to activated (right)]. The proton pump (H+/K+-ATPase) moves to the secretory canaliculus (plasma membrane) from cytoplasmic tubulovesicles. H2-blockers compete with histamine at the histamine H2-receptor. (B) Ion balance in the parietal cell. The H+ transported by the proton pump are supplied by carbonic anhydrase. Bicarbonate, the other product of this enzyme, is antiported with Cl- and Cl- is secreted through a Cl- channel. The potassium (K+) imported by the proton pump are again excreted by a K+ channel. The proton pump has catalytic α- and glycosylated β-subunits. The drug omeprazole covalently modifies cysteine residues located in the extracytoplasmic domain of the α-subunit and inhibits the proton pump. Thick arrows indicate the binding sites for inhibitors. |
| page 90 | | | page 91 |
| VARIOUS DRUGS INHIBIT TRANSPORTERS IN MUSCLE |
| Phenylalkylamine (verapamil), benzothiazepine (diltiazem), and dihydropyridine (DHP; nifedipine) are Ca2+-channel blockers that inhibit VDCCs (Fig. 7.6). Ryanodine inhibits the Ca2+-release channel in the sarcoplasmic reticulum. These drugs are used to inhibit both the increase in cytoplasmic Ca2+ concentration and the force of muscle contraction. In contrast, cardiac glycosides such as ouabain and digoxin increase heart muscle contraction and are used for treatment of congestive heart failure. They act by inhibiting the Na+/K+-ATPase that generates the Na+ concentration gradient used to drive export of Ca2+ by the Na+/Ca2+ antiporter. Snake venoms such as α-bungarotoxin, and tetrodotoxin from the puffer fish inhibit voltage-dependent Na+ channels. Lidocaine, an Na+-channel blocker, is used as a local anesthetic and antiarrhythmic drug. Inhibition of Na+ channels represses transmission of the depolarization signal. |
| INHIBITING THE GASTRIC PROTON PUMP AND ERADICATION OF HELICOBACTER PYLORI |
| Chronic strong acid secretion by the gastric proton pump injures the stomach and the duodenum, leading to gastric and duodenal ulcers. Proton pump inhibitors such as omeprazole are delivered to parietal cells from the circulation after oral administration. Omeprazole is a prodrug: it accumulates in the acidic compartment, as it is a weak base, and is converted to the active compound under the acidic conditions in the gastric lumen. The active form covalently modifies cysteine residues located in the extracytoplasmic domain of the proton pump. H2-blockers (receptor antagonists) such as cimetidine and ranitidine indirectly inhibit acid secretion by competing with histamine for its receptor (Fig. 7.8). |
| Comment. Infection of the stomach by Helicobacter pylori also causes ulcers and is associated with an increased risk of gastric adenocarcinoma. Recently, antibiotic treatment has been introduced to eradicate H. pylori. Interestingly, antibiotic treatment together with omeprazole is much more effective, possibly because of an increased stability of the antibiotic under the weakly acidic condition produced by proton pump inhibition. |
|