| THE PYRUVATE DEHYDROGENASE COMPLEX (PDC)
|
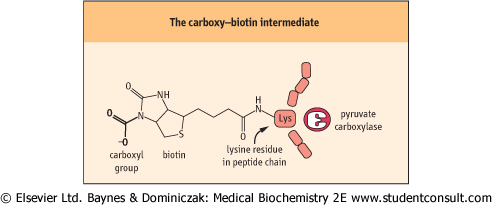
| Figure 13.4 The carboxy-biotin intermediate. Pyruvate carboxylase catalyzes carboxylation of pyruvate to oxaloacetate. The coenzyme, biotin, is covalently bound to pyruvate carboxylase, and transfers the carbon originating from CO2 to pyruvate. |
The pyruvate dehydrogenase complex (PDC) serves as a bridge between carbohydrates and the TCA cycle (Fig. 13.5).
PDC is one of several α-ketoacid dehydrogenases having analogous reaction mechanisms, including α-ketoglutarate dehydrogenase in the TCA cycle and α-ketoacid dehydrogenases associated with the catabolism of leucine, isoleucine and valine. Its irreversibility explains in part why acetyl-CoA cannot yield a net synthesis of glucose . The complex functions as a unit consisting of three principal enzymes: . The complex functions as a unit consisting of three principal enzymes:
- pyruvate dehydrogenase (PDH; E1)
- dihydrolipoyl transacetylase (E2)
- dihydrolipoamide dehydrogenase (E3)
|
| Two additional enzymes of the complex, pyruvate dehydrogenase kinase and pyruvate dehydrogenase phosphatase, regulate its activity by covalent modification via reversible phosphorylation/dephosphorylation. There are four known isoforms of the kinase, and two of the phosphatase; the relative amounts of each are cell-specific. Intermediates traverse minimal distances between each catalytic step, because they are tethered to the complex during the reaction sequence (see Figs 13.5 and 13.6).
|
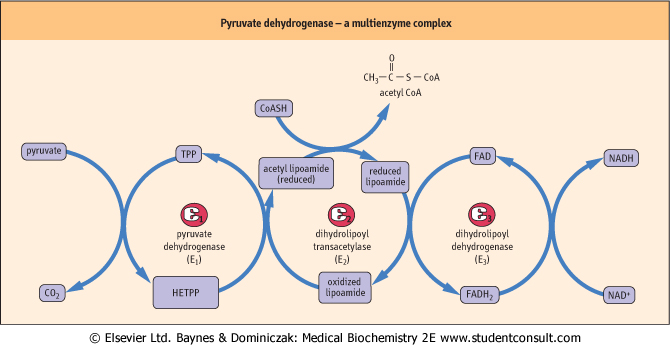
| Figure 13.5 Mechanism of action of the pyruvate dehydrogenase complex. The three enzyme components of the pyruvate dehydrogenase complex are pyruvate dehydrogenase (E1), dihydrolipoyl transacetylase (E2) and dihydrolipoyl dehydrogenase (E3). Pyruvate is first decarboxylated by the thiamine pyrophosphate-containing enzyme (E1), forming CO2 and hydroxyethyl-thiamine pyrophosphate (HETPP). Lipoamide, the prosthetic group on E2, serves as a carrier in the transfer of the 2-carbon unit from HETPP to coenzyme A (CoA). The oxidized, cyclic disulfide form of lipoamide accepts the hydroxyethyl group from HETPP. The lipoamide is reduced and the hydroxyethyl group converted to an acetyl group during this transfer reaction, forming acetyldihydrolipoamide. Following transfer of the acetyl group to CoA, E3 reoxidizes the lipoamide, using FAD, and the FADH2 is, in turn, oxidized by NAD+, yielding NADH. |
| page 178 |  | | page 179 |
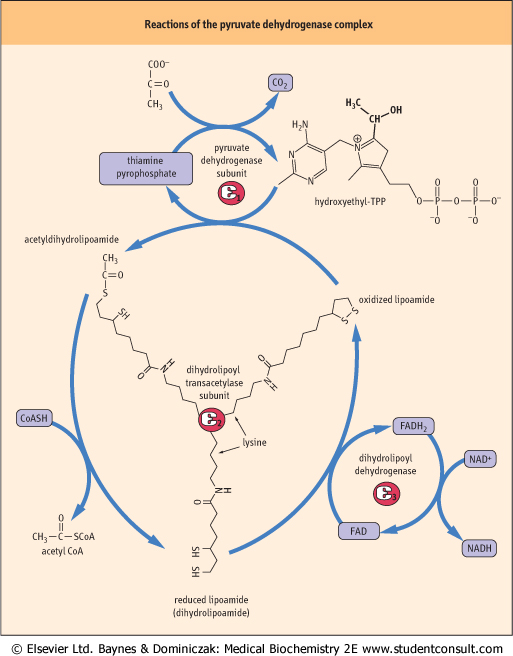
| Figure 13.6 Lipoic acid in the pyruvate dehydrogenase complex. The coenzyme lipoamide is attached to a lysine residue in the transacetylase subunit of pyruvate dehydrogenase. Lipoamide moves from one active site to another on the transacetylase subunit in a 'swinging arm' mechanism. The structures of thiamine pyrophosphate (TPP) and lipoamide are shown. |
| PYRUVATE DEHYDROGENASE COMPLEX DEFICIENCY |
| Most children with this enzyme deficiency present in infancy with delayed development and reduced muscle tone often associated with ataxia and seizures. Some infants have congenital malformations of the brain. |
| Comment. Without mitochondrial oxidation, pyruvate is reduced to lactate. The ATP yield from anaerobic glycolysis is less than a tenth of that produced from complete oxidation of glucose via the tricarboxylic acid cycle. The diagnosis is suggested by elevated lactate, but with a normal lactate/pyruvate ratio, i.e. no evidence of hypoxia. A ketogenic diet and severe restriction of protein (<15%) and carbohydrate (<5%) improves mental development. Such treatment ensures that the cells use acetyl-CoA from fat metabolism. A few children show a reduction in plasma lactate on treatment with large doses of thiamine, but the outlook is generally poor. |
| page 179 | | | page 180 |
| Five coenzymes are required for PDC activity: thiamine pyrophosphate, lipoamide (lipoic acid bound in amide linkage to protein), CoA, FAD, and NAD+. For their synthesis, thiamin,
pantothenic acid, riboflavin and nicotinamide are required. Deficiencies in any of these vitamins have obvious effects on energy metabolism. For example, increases in cellular concentrations of pyruvate and α-ketoglutarate are found in beri-beri because of thiamin deficiency (Chapter 10). In this case, all of the proteins are available, but the relevant coenzyme is not, and the conversions of pyruvate to acetyl-CoA and α-ketoglutarate to succinyl-CoA are significantly reduced. Symptoms include cardiac and skeletal muscle weakness and neurologic disease. Thiamin deficiency is common in alcoholism, because distilled spirits are devoid of vitamins, and symptoms of beri-beri are often observed.
|
|