| Mitochondrial β-oxidation
|
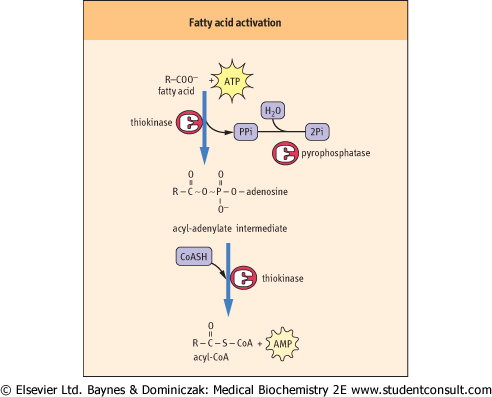
Figure 14.1 Activation of fatty acids by fatty acyl-CoA synthetase (thiokinase). ATP forms an enzyme-bound acyl adenylate intermediate, which is discharged to form acyl CoA. AMP, adenosine monophosphate; CoASH, coenzyme A; PPi, inorganic pyrophosphate. monophosphate; CoASH, coenzyme A; PPi, inorganic pyrophosphate. |
|
Table 14-1.
Metabolism of the four classes of fatty acids. |
| Body_ID: None |
| Metabolism of fatty acids |
| Body_ID: T014001.50 |
| Size class | Number of carbons | Site of catabolism | Membrane transport |
| Body_ID: T014001.100 |
| Short-chain | 2-4 | mitochondrion | diffusion |
| Body_ID: T014001.150 |
| Medium-chain | 4-12 | mitochondrion | diffusion |
| Body_ID: T014001.200 |
| Long-chain | 12-20 | mitochondrion | carnitine cycle |
| Body_ID: T014001.250 |
| Very-long-chain | >20 | peroxisome | unknown |
| Body_ID: T014001.300 |
| page 190 |  | | page 191 |
| Deficiencies in carnitine metabolism |
| The clinical presentation of deficient carnitine metabolism occurs in infancy and is often life threatening. Characteristic features include hypoketotic hypoglycemia, hyperammonemia, and altered plasma free carnitine concentration. Hepatic damage, cardiomyopathy, and muscle weakness are common. |
| Comment. Carnitine is synthesized from lysine and α-ketoglutarate, primarily in liver and kidney, and is normally present in plasma in a concentration of about 50 mmol/L (0.8 mg/dL). There are high-affinity uptake systems for carnitine in most tissues, including the kidney, which resorbs carnitine from the glomerular filtrate, limiting its excretion in urine. Homozygous deficiencies in carnitine transport, CPTs-I and -II, and the translocase result in defects in long-chain fatty acid oxidation. Plasma and tissue carnitine concentrations decrease to <1 mmol/L in carnitine transport deficiency, because of both defective uptake into tissues and excessive loss in urine. On the other hand, plasma free carnitine may exceed 100 mmol/L (2 mg/dL) in CPT-I deficiency. In both translocase and CPT-II deficiency, total plasma carnitine may be normal, but is mostly in the form of acyl carnitine esters of long-chain fatty acids - in the former case, because they cannot be transported into the mitochondrion, and in the latter because of backflow out from mitochondria. These diseases are treated by carnitine supplementation, by frequent high-carbohydrate feeding, and by avoidance of fasting. |
| Fatty acyl-CoAs are oxidized in a cycle of reactions involving oxidation of the β-carbon to a ketone; hence the terminology,
β-oxidation (Figs 14.3 and 14.4). The oxidation is followed by cleavage between the α- and β-carbons by a thiolase reaction. One mole each of acetyl-CoA, FADH2, and NADH + H+ is formed during each cycle, along with a fatty acyl-CoA with two fewer carbon atoms. For a 16-carbon fatty acid, such as palmitate, the cycle is repeated seven times, yielding 8 moles of acetyl-CoA (Fig. 14.3, Table 14.1), plus seven moles of FADH2 and 7 moles of NADH + H+. This process occurs in
the mitochondrion, and the reduced nucleotides are used directly for synthesis of ATP by oxidative phosphorylation (Table 14.2).
|
|
Table 14-2.
Comparative energy yield from glucose and palmitate. |
| Body_ID: None |
| Caloric value of glucose and palmitate |
| Body_ID: T014002.50 |
| Substrate | Molecular weight | Net ATP yield (mol/mol) | ATP (mol/g) | Caloric value Cal/g (kJ) |
| Body_ID: T014002.100 |
| glucose | 180 | 36-38 | 0.2 | 4 (17) |
| Body_ID: T014002.150 |
| palmitate | 256 | 129 | 0.5 | 9 (37) |
| Body_ID: T014002.200 |
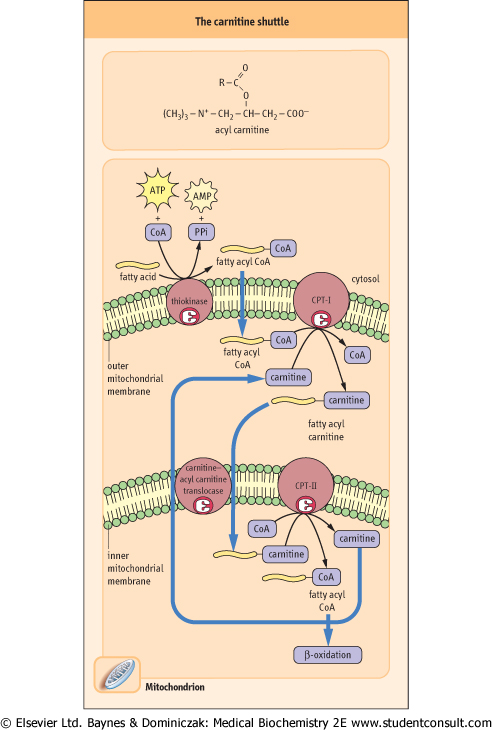
| Figure 14.2 Transport of long-chain fatty acids into the mitochondrion. The three components of the carnitine pathway include extra- and intra-mitochondrial CPTs and the carnitine-acyl carnitine translocase. |
| page 191 | | | page 192 |
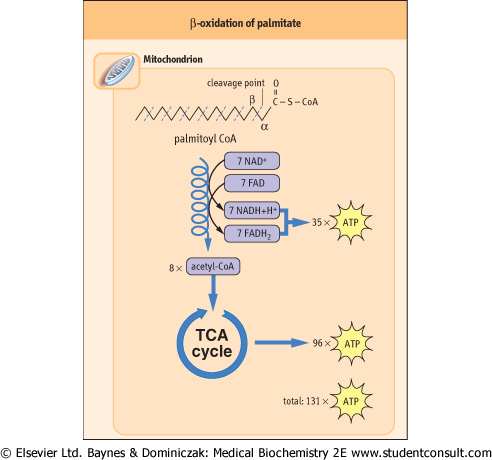
| Figure 14.3 Overview of β-oxidation of palmitate. In a cycle of reactions, the carbons of the fatty-acyl-CoA are released in two-carbon acetyl-CoA units; the yield of 35 ATP from this β-oxidation is nearly equivalent to that from complete oxidation of glucose. In liver, the acetyl CoA units are then used for synthesis of ketone bodies, and in other tissues they are metabolized in the TCA cycle to form ATP. The complete oxidation of palmitate yields a net 129 moles of ATP, after correction for the 2-mole equivalents of ATP invested at the thiokinase reaction. The overall production of ATP per gram of palmitate is about twice that per gram of glucose, because glucose is already partially oxidized in comparison with palmitate. For this reason, as illustrated in Table 14.2, the caloric value of fats is about twice that of sugars. |
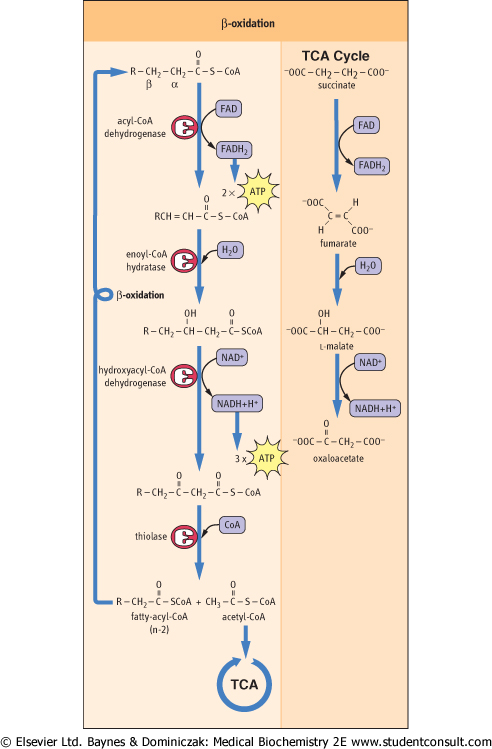
| The four steps in the cycle of β-oxidation are shown in detail in Figure 14.3. Note the similarity between the sequence of these reactions and those between succinate and oxaloacetate in the TCA cycle. In common with succinate dehydrogenase, acyl-CoA dehydrogenase uses the prosthetic group FAD as a coenzyme, and is an integral protein in the
inner mitochondrial membrane. Even the trans geometry of fumarate and the stereochemical configuration of l-malate in the TCA cycle are mirrored by the trans geometry of the trans-enoyl-CoA and l-hydroxyacyl-CoA intermediates in β-oxidation. The last step of the β-oxidation cycle is catalyzed by thiolase, which traps the energy obtained from the carbon-carbon bond cleavage as acyl-CoA, allowing the cycle to continue without the necessity of reactivating the fatty acid. The cycle continues until all the fatty acid has been converted to acetyl-CoA, the common intermediate in the oxidation of carbohydrates and lipids.
|
| Figure 14.4 β-Oxidation of fatty acids. Oxidation occurs in a series of steps at the carbon that is β to the keto group. Thiolase cleaves the resultant β-ketoacyl-CoA derivative to give acetyl-CoA and a fatty acid with two fewer carbon atoms, which then re-enters the β-oxidation cascade. Note the similarity between these reactions and those of the TCA cycle, shown on the right. |
| page 192 | | | page 193 |
| IMPAIRED OXIDATION OF MEDIUM-CHAIN FATTY ACIDS |
| Fatty acyl CoA dehydrogenase |
| Fatty acyl CoA dehydrogenase is not a single enzyme, but a family of enzymes with chain-length specificity for oxidation of short-, medium- and long-chain fatty acids; fatty acids are transferred from one enzyme to the other during chain-shortening β-oxidation reactions. Medium-chain fatty acyl CoA dehydrogenase (MCAD) deficiency is an autosomal recessive disease characterized by hypoketotic hypoglycemia. It presents in infancy, and is characterized by high concentrations of medium-chain carboxylic acids, acyl carnitines, and acyl glycines in plasma and urine. Hyperammonemia may also be present, as a result of liver damage. Concentrations of hepatic mitochondrial medium-chain acyl CoA derivatives are also increased, limiting β-oxidation and recycling of CoA during ketogenesis. The inability to metabolize fats during fasting is life threatening because it limits gluconeogenesis and causes hypoglycemia. MCAD deficiency is treated by frequent feeding, avoidance of fasting, and carnitine supplementation. Deficiencies in short- and long-chain fatty acid dehydrogenases have also been described, and have similar clinical features. |
|