| METABOLISM OF LIPOPROTEINS
|
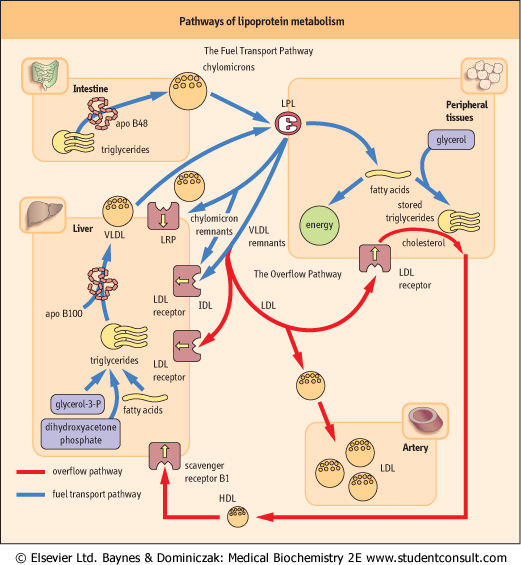
The stages of lipoprotein metabolism are as follows:
- Assembly of lipoprotein particles. Chylomicrons are assembled in the intestine, and VLDL in the liver;
- Transfer of fatty acids from lipoproteins to cells. This facilitated by enzymes, LPL and HTGL and transforms chylomicrons and VLDL into the relevant remnant particles (remnants);
- Binding of the remnant particles to membrane receptors and their cellular uptake;
- Transformation of some remnants into LDL, their subsequent receptor binding and cellular uptake.
- Reverse cholesterol transport, i.e. removal of cholesterol from cells by HDL particles, its esterification by LCAT and either transport back to the liver, or CETP-mediated transfer to other lipoproteins by which they re-enter the VLDL-IDL-LDL pathway.
|
| Chylomicrons transport dietary fat
|
| Triacylglycerols contained in food are first acted upon by pancreatic lipases and are absorbed as monoacylglycerols, free fatty acids, and free glycerol (see Chapter 9). In the intestinal cells (enterocytes), triacylglycerols are resynthesized and, together with phospholipids, cholesterol and apoB48, are reassembled into chylomicrons. These are secreted into the lymph and reach plasma through the thoracic duct. They normally appear in plasma only after fat-containing meals, giving plasma a milky appearance. In the peripheral tissues, such as muscle and adipose tissue, chylomicron triacylglycerols are hydrolyzed by LPL. The remaining smaller particles are the chylomicron remnants. The remnants travel to the liver, where they bind to the LDL receptor and the LRP. The half- life of chylomicrons in plasma is less than 1 h (Fig. 17.6).
|
| VLDL transport endogenously synthesized triglycerides
|
| Endogenously synthesized triacylglycerols are transported by VLDL, which are assembled in the liver. There, VLDL are 'built up' around apoB100 molecules. The binding of triacylglycerols to apoB is facilitated by the microsomal triglyceride transfer protein (MTP). Unused ApoB100 is degraded by ubiquitin-dependent protease (see Chapter 32, Fig. 32.10).
|
| In plasma, VLDL acquire cholesteryl esters and apoproteins (including apoE) from HDL. Its triacylglycerols are hydrolyzed by LPL in a way analogous to chylomicrons, and this yields VLDL remnants. At this stage apoE assumes its receptor-binding conformation and it binds to the apoB/E receptor. Most of the remnants are taken up by the liver. The remaining ones are further hydrolyzed by HTGL to yield IDL, which in turn, by losing still more triacylglycerol, transform into LDL. By this stage, all apoproteins except for the apoB100 had been shed from the particle.
|
| LDL is taken up by cells by the same route as remnant particles
|
| page 231 |  | | page 232 |
| Figure 17.6 Lipoprotein metabolism: the fuel transport pathway and the overflow pathway. The fuel transport pathway: chylomicrons transport triglyceride to the periphery, and their remnants are metabolized in the liver. VLDL transport fuel from the liver to peripheral tissues, and the remnants also return to the liver. Part of VLDL remnants and IDL are further converted into LDL, which enter the overflow pathway. The overflow pathway: LDL travel in blood from the liver through the peripheral tissues and back to the liver. On its way it may enter the arterial wall. The amount of lipids deposited in the arteries is proportional to their plasma concentration. Cholesterol is removed from cells and transported back to the liver by HDL particles. LPL: lipoprotein lipase, LRP: LDL-receptor-related protein. |
| By shedding almost all triacylglycerols, LDL becomes relatively rich in cholesterol. The apoB100 now acquires a
receptor-binding conformation and begins to control LDL internalization. LDL are taken up by the apoB/E receptor either in the liver (approximately 80% of particles follow this path) or in the peripheral tissues (see Fig. 17.6). Interestingly the apoB-containing LDL particles have lower affinity toward the receptor than apoE-containing remnants.
|
| A sophisticated system controls intracellular cholesterol synthesis and uptake
|
| After internalization, the LDL-receptor complex is digested by lysosomal enzymes and the released cholesterol is esterified within the cell by SOAT. The receptor recycles back to the membrane. The free cholesterol released within the cell regulates the rate of its own endogenous synthesis. The key element in the regulation of intracellular cholesterol concentration are the sterol regulatory element-binding proteins (SREBPs). SREBPs are a family of transcription factors. They regulate the transcription of genes encoding enzymes responsible for cholesterol synthesis such as 3-hydroxy-3-methylglutaryl coenzyme A synthase and HMG-CoA reductase, and also the gene coding for the apoB/E (LDL) receptor (see also Chapter 16). Thus, depletion of hepatic sterols increases SREBP level, increasing both the cholesterol synthesis, and the expression of LDL receptor. On the other hand, increased intracellular cholesterol concentration inhibits the SREBP pathway and thus represses the receptor gene. It also decreases cholesterol synthesis.
|
| page 232 | | | page 233 |
| SREBPs are synthesized as integral membrane proteins in a precursor form. SREBP is activated by another protein, the SREBP cleavage-activating protein (SCAP) which has a sterol-sensing domain. Thus, when cell cholesterol concentration is low, SCAP moves together with the membrane-embedded SREBP precursor from the endoplasmic reticulum to the cleaving enzyme (a serine protease) located in the Golgi apparatus. There, the protease cleaves the SREBP precursor and
liberates the active transcription factor, which translocates to the nucleus (see also Chapter 16).
|
| Cholesterol is removed from cells by the reverse cholesterol transport
|
| Human beings cannot metabolize the cholesterol ring: it needs to be transported to the liver and excreted either in a free form, or as a bile acid. The system that allows the removal of cholesterol from cells is known as the reverse cholesterol transport. It is mediated by the HDL.
|
| HDL are formed in the liver and intestine as discoidal, lipid-poor particles (pre-β HDL) which contain mainly apoAI: they are partly constructed from the excess phospholipid shed from VLDL during its hydrolysis by LPL. These nascent HDL accept cholesterol from cells.
|
| A membrane protein known as cholesterol efflux regulatory protein (CERP, also called the ATP-binding cassette transporter A1 - see Chapter 7) plays a major role in cholesterol efflux from cells. It uses ATP as a source of energy, CERP is rate-limiting in the efflux of free cholesterol to apoAI. Other ATP-binding cassette transporters, known as ABCG5 and ABCG8, are also involved in cholesterol transport; they reside in the apical membrane of the hepatocyte where they control the movement of cholesterol into bile. The role of CERP in cholesterol efflux was defined as a result of the study of extremely rare dyslipidemia called Tangier disease. Patients suffering from Tangier disease have almost no HDL, and show accumulation of cholesteryl esters in tissues such as tonsils (which acquire a characteristic orange colour), and also in the spleen, intestinal mucosa, and cornea. Patients with Tangier disease suffer from coronary disease more frequently than those without.
|
| After the nascent HDL acquires the free cholesterol, it is esterified by LCAT; the cholesteryl esters move deeper into the HDL particle, and the particle assumes a spherical shape. It is now known as HDL-3. Subsequently, aided by cholesterol ester transfer protein, HDL transfers part of its cholesteryl esters to triglyceride-rich lipoproteins in exchange for triglycerides. At that point it gets even larger, and is now designated HDL-2. This particle now binds to the class B scavenger receptor in the liver. However, binding does not result in HDL internalization, but only in the transfer of cholesterol to the cell membrane. The particle shrinks again, and some of its redundant parts become nascent HDL and participate in the next cycle of transport (Fig. 17.7).
|
| Cholesterol delivered to the liver by the HDL is excreted either as a free sterol or as bile acids. This is controlled by the sterol-responsive transcription factors, termed the liver X receptors (LRX). These receptors form dimers with another class of intracellular receptors, the retinoid X receptors (RXRs) and control the expression of several proteins such as transfer proteins and the enzyme 7α-hydroxylase (CYP7A1), which plays a key role in the biliary cholesterol excretion (see Chapter 16).
|
| PEROXISOME PROLIFERATION ACTIVATING RECEPTORS (PPARS) ARE TRANSCRIPTION FACTORS INVOLVED IN CONTROL OF LIPOPROTEIN METABOLISM AND IN INSULIN ACTION |
PPARs are lipid-activated transcription factors (Chapter 33) that regulate the genes controlling lipid and glucose homeostasis. PPARs, similarly to the liver X receptors, form dimers with retinoid X receptor RXR; it is this dimer that subsequently binds to PPAR-response elements. homeostasis. PPARs, similarly to the liver X receptors, form dimers with retinoid X receptor RXR; it is this dimer that subsequently binds to PPAR-response elements. |
| There are three PPAR proteins: PPARα, PPAR β/δ and PPARγPPARα is present in liver, kidney, heart, and skeletal muscle and is involved in the control of fatty acid and lipoprotein metabolism. PPARγis present in the brown and white adipose tissue, and affects cell differentiation, insulin action and fatty acid synthesis. It also stimulates triacylglycerol synthesis and storage. PPARβ/δparticipates in brain lipid metabolism and affects proliferation and differentiation of adipocytes. PPARs also regulate reverse cholesterol transport and bile acid synthesis. For instance, PPARαregulates the expression of the AI-CIII-AIV gene cluster; this increases the expression of apoAI and apoAII and reduces the expression of apoCIII gene. It also increases the expression of LPL. |
| PPARs are important targets of drug action. The commonly used lipid-lowering drugs, derivatives of fibric acid (fibrates) activate PPARα. New antidiabetic drugs, thiazolidinediones, increase insulin sensitivity in insulin-resistant patients (see Chapter 20) activate PPARγ. |
|