| page 257 |  | | page 258 |
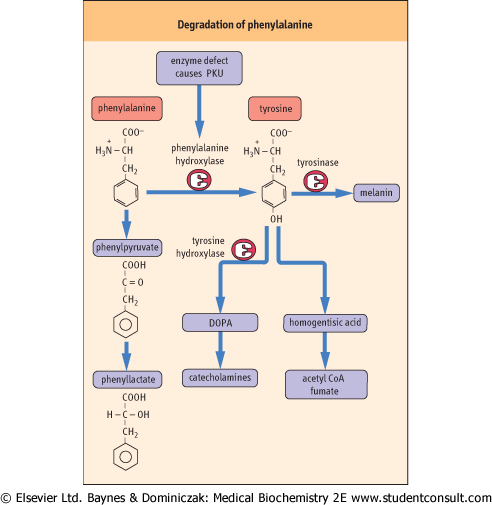
| Figure 18.12 Degradation of phenylalanine. In order to enter normal metabolism, phenylalanine must be hydroxylated by the enzyme, phenylalanine hydroxylase. A defect in this enzyme leads to phenylketonuria (PKU). Tyrosine is a precursor of acetyl-CoA and fumarate, catecholamine hormones, the neurotransmitter dopamine, and the pigment melanin. DOPA: dihydroxyphenylalanine. |
| Phenylketonuria (PKU) results from a deficiency of the enzyme, phenylalanine hydroxylase. The hydroxylation of phenylalanine is a required step in both the normal degradation of the carbon skeleton of this amino acid and the synthesis of tyrosine (Fig. 18.12). When untreated, this metabolic defect leads to excessive urinary excretion of phenylpyruvate and phenyllactate, and severe mental retardation. In addition, individuals with PKU tend to have very light skin pigmentation, unusual gait, stance, and sitting posture, and a high frequency of epilepsy. In the USA, this autosomal recessive defect occurs in about 1 in 30 000 live births. Because of this, and the ability to prevent the most serious consequences of the defect by a low-phenylalanine diet, newborns in most developed countries are routinely tested for blood concentrations of phenylalanine. Fortunately, with early detection and the use of a diet restricted in phenylalanine but supplemented with tyrosine, most of the mental retardation can be avoided. Mothers who are homozygous for this defect have a very high probability of bearing children with congenital defects and mental retardation unless their blood phenylalanine concentrations can be controlled by diet. The developing fetus is very sensitive to the toxic effects of high concentrations of phenylalanine and
related phenylketones. Not all hyperphenylalaninemias are caused by a defect in phenylalanine hydroxylase. In some cases, there is a defect in biosynthesis or reduction of a required tetrahydrobiopterin cofactor.
|
|