| Tropomyosin and troponins
|
| Tropomyosin stabilizes F-actin and coordinates conformational changes among actin subunits during contraction
|
| Tropomyosin is a fibrous protein that extends along the grooves of F-actin, each molecule contacting about seven G-actin subunits. Tropomyosin has a role in stabilizing F-actin and coordinating conformational changes among actin subunits during contraction. In the absence of Ca2+, tropomyosin blocks the myosin binding site on actin. A complex of troponin proteins is bound to tropomyosin: Tn-T (tropomyosin-binding), Tn-C (calcium-binding) and Tn-I (inhibitory subunit). Troponins modulate the interaction between actin and myosin. Calcium binding to Tn-C, a calmodulin-like protein, induces changes in Tn-I, which are then transduced to tropomyosin, moving it out of the myosin-binding site and permitting actin-myosin interactions.
|
| The sliding-filament model of muscle contraction
|
| The general features of the sliding-filament model of muscle contraction are described by a series of chemical and structural changes in the actomyosin complex. The contractile response is powered by reversible 'cross-bridge' interactions between the myosin head and its actin-binding site. A cycle of binding of myosin to actin, conformational changes in the hinge regions of myosin, and release of myosin occurs, with the conformational change providing the 'power stroke' for muscle contraction. This sequence of reactions is summarized in Figure 19.4. The conformational change in the hinge regions of myosin is induced by hydrolysis of ATP and relaxed by dissociation of ADP and Pi. The latter process, the dissociation of ADP and Pi, rather than the hydrolysis of ATP, is the rate-limiting step in myosin ATPase activity and muscle contraction. The stability of the contracted state is maintained by multiple and continuous actin-myosin interactions, so that slippage is minimized until calcium is removed from the sarcoplasm, allowing the muscle to relax.
|
| Excitation-contraction coupling: The calcium trigger
|
| DUCHENNE MUSCULAR DYSTROPHY |
| A young boy was brought to the clinic because his mother had noticed that he walked with a waddling gait. Physical evaluation confirmed muscle weakness especially in the legs although his calf muscles were large and firm. There was a 20-fold elevation in serum creatine (phospho) kinase (CK) activity, identified as the MM (muscle) isozyme. Histology revealed muscle loss, some necrosis, and increased connective tissue and fat volume in muscle. A tentative diagnosis of Duchenne muscular dystrophy (DMD) was confirmed by immunoelectrophoretic (Western blot) analysis showing the lack of the cytoskeletal protein dystrophin in muscle. |
| Comment. Dystrophin is a high-molecular-weight cytoskeletal protein that reinforces the plasma membrane of the muscle cell and mediates interactions with the extracellular matrix. In its absence, the plasma membrane of muscle cells is damaged during the contractile process, leading to muscle cell death. The dystrophin gene is located on the X-chromosome and is unusually long, nearly 2.5 × 106 base pairs. Mutations are relatively common, the frequency of DMD being approximately 1 in 3500 male births. DMD is a progressive myodegenerative disease, commonly leading to confinement to a wheelchair by puberty, with death by age 20 years from respiratory or cardiac failure. Dystrophin is completely absent in DMD patients. A variant of the disease, known as Becker muscular dystrophy, has milder symptoms and is characterized by expression of an altered dystrophin protein and survival into the fourth decade. Although there is currently no treatment, the injection of satellite cells (myogenic stem cell) that express dystrophin protein and incorporate into the dystrophic skeletal muscle has shown promise in animal trials. |
| page 265 |  | | page 266 |
|
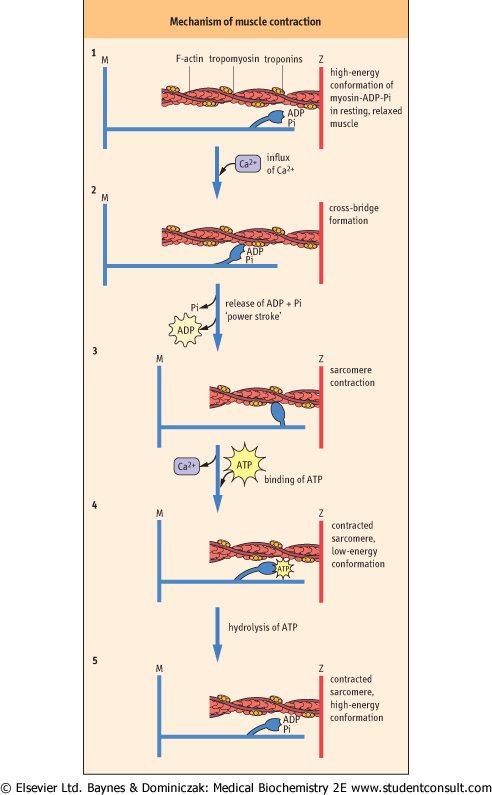
Figure 19.4 Proposed stages in muscle contraction, according to the sliding-filament model.
- (1) In resting, relaxed muscle, calcium concentration is ∼10-7 mol/L. The head group of myosin chains contains bound ADP and Pi, and is extended forward along the axis of the myosin helix in a high-energy conformation. Although the myosin-ADP-Pi complex has a high affinity for actin, binding of myosin to actin is inhibited by tropomyosin, which blocks the myosin-binding site on actin at low calcium concentration.
- (2) When muscle is stimulated, calcium enters the sarcoplasm through voltage-gated calcium channels (see Chapter 7). Calcium binding to Tn-C causes a conformational change in Tn-I, which is transmitted through Tn-T to tropomyosin. Movement of tropomyosin exposes the myosin-binding site on actin. Myosin-ADP-Pi binds to actin, forming a cross-bridge.
- (3) Release of Pi, then ADP, from myosin during the interaction with actin is accompanied by a major conformational change in myosin, producing the 'power stroke', which moves the actin chain about 10 nm (100 Å) in the direction opposite the myosin chain, increasing their overlap and causing muscle contraction.
- (4) The uptake of calcium from the sarcoplasm and binding of ATP to myosin leads to dissociation of the actomyosin cross-bridge.
- (5) The ATP is hydrolyzed, and the free energy of hydrolysis of ATP is conserved as the high-energy conformation of myosin, setting the stage for continued muscle contraction in response to the next surge in Ca2+ concentration in the sarcoplasm.
|
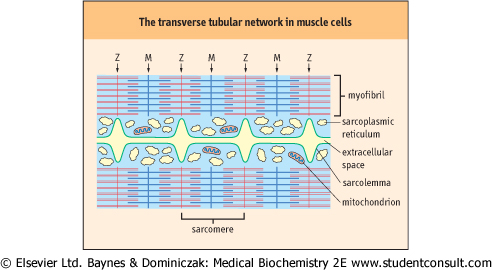
| Figure 19.5 Side-view of the transverse tubular network in skeletal muscle cells. Transverse tubules are invaginations of the sarcolemma, which are in intimate contact with the sarcoplasmic reticulum (SR). The SR is a continuous, tubular compartment in close association with the myofibrils. The transverse tubules are extensions of the sarcolemma around the Z-line. They transmit the depolarizing nerve impulse to terminal regions of the SR, coordinating calcium release and contraction of the myofibril. |
| page 266 | | | page 267 |
| Sarcopenia is defined as the loss of skeletal muscle mass with age. Sarcopenia is accelerated in humans after the fifth decade of life and can lead to frailty and loss of functional capacity. Besides the basic erosion of quality of life, loss of skeletal muscle mass also increases the risk of mortality and morbidity. The cause of sarcopenia appears to be related to both a biological program of muscle fiber loss and decreased physical activity. Muscle fiber innervation by spinal motor neurons is critical to both development and maintenance of the mature muscle phenotype. Spinal motor neurons decrease in number with advancing age, possibly because of cumulative oxidative damage to these post-mitotic cells. The loss of motor neurons induces muscle fiber loss and an increase in existing motor unit size, which decreases fine motor skill. Sarcopenia has also been linked to age-induced systemic changes to the endocrine, cardiovascular, and immune systems, whose functions are all critical for the maintenance of skeletal muscle mass. |
| Comment: The scientific evidence is clear that most elder individuals can increase muscle strength and mass with a regular resistance exercise program. Pharmaceutical treatments have also been examined for individuals who cannot regularly exercise. Currently there is no treatment for spinal motor neuron loss. Pharmaceutical treatments targeting muscle have had varying degrees of success, but are usually limited by side-effects. The treatments include endocrine interventions with male or female sex hormone replacement therapy, and growth hormone therapy. Anti-inflammatory medication is also employed to allow individuals to participate in physical activity programs. One of the best defenses from sarcopenia may be regular exercise in order to maintain muscle mass during middle age. |
|
| The calcium content of the muscle cytoplasm (sarcoplasm) is normally very low, 10-7 mol/L or less, but increases rapidly by ∼100-fold in response to neural stimulation, leading to ATP hydrolysis and muscle contraction. Muscle cells have an adapted smooth endoplasmic reticulum organelle, known as the sarcoplasmic reticulum (SR), which serves as the site of calcium sequestration inside the cell. The plasma membrane of the myofiber cell, known as the sarcolemma, invaginates in and around the myofibrils at the Z-lines, forming a series of transverse tubules that indirectly interact with the SR (Fig. 19.5). The muscle cell responds to motor nerve stimulation by initiating a wave of depolarization of the Na+/K+-gradient across the muscle plasma membrane, which is transmitted to the SR, causing a voltage-gated opening of SR Ca2+-channels. Calcium is released from the SR, and the influx of Ca2+ into the muscle cytoplasm (sarcoplasm) triggers muscle contraction. In striated muscle, calcium interacts with troponin-C on the thin filament to allow actin-myosin interaction. Muscle contraction is mediated by changes in the conformations and
interactions of actin and myosin. The actomyosin complex thereby transforms the chemical energy of ATP into the mechanical action of muscle.
|
| Troponin protein is not expressed in smooth muscle. In this case, calcium triggers contraction by calmodulin binding. Calcium-calmodulin binding activates myosin light chain kinase. These events lead to myosin phosphorylation, which allows myosin-actin interaction.
|
| Although muscle contraction is triggered by increased calcium, muscle relaxation is dependent on calcium being pumped back into the SR. The rate of muscle relaxation is directly related to SR-calcium ATPase activity. The SR is rich in a calcium-ATPase, which pumps calcium into the SR, maintaining cytosolic calcium in the muscle cell at submicromolar (∼10-7 mol/L) concentrations. At the same time, the concentration of calcium in the SR is in the mmol/L range, comparable to that in the plasma compartment.
|
Metabolic syndrome (Syndrome X), characterized by insulin resistance, hyperinsulinemia, hypertension, and hyperlipidemia, is a major, age-dependent risk factor for diabetes and cardiovascular disease. Loss of muscle mass and insulin sensitivity can dramatically influence the progression from Syndrome X to frank disease through their effect on blood glucose concentration. A half-hour of vigorous physical activity can increase skeletal muscle glucose uptake for up to 24 hours after the exercise period. Regular exercise also reduces the loss of muscle mass, improves muscle perfusion, and enhances the insulin sensitivity of muscle. There is clear evidence that obese people also benefit from regular exercise (see Chapters 20 and 21). concentration. A half-hour of vigorous physical activity can increase skeletal muscle glucose uptake for up to 24 hours after the exercise period. Regular exercise also reduces the loss of muscle mass, improves muscle perfusion, and enhances the insulin sensitivity of muscle. There is clear evidence that obese people also benefit from regular exercise (see Chapters 20 and 21). |
| Muscle: An excitable tissue
|
| Muscle has the ability to depolarize upon neural stimulation, and this depolarization is critical for the calcium release that triggers contraction. However, skeletal, cardiac, and smooth muscle receive this neural depolarization in different manners, and have different structural adaptations at the sarcolemma to account for depolarization propagation. Skeletal muscle contraction is volitional and fibers are innervated by motor nerve endplates that originate in the spinal cord. The neuromuscular junction is a special structural feature of skeletal muscle that is not found in cardiac or smooth muscle. Each individual fiber is innervated by only one motor nerve, and all the fibers innervated by one nerve are defined as a motor unit. Motor unit control and synchronization is the basis for coordinated whole muscle contraction.
|
| Cardiac muscle is striated and contracts rhythmically under involuntary control. The general mechanism of contraction of heart muscle is similar to that in skeletal muscle, but the SR is less developed, and the transverse tubule network, an extension of the plasma membrane, is more developed in the heart. Thus, the heart is more dependent on, and actually requires, extracellular calcium for its contractile response. Lacking direct neural contact, cardiac myocytes propagate depolarization from a single node, the SA-node, throughout the myocardium. The depolarization is passed cell to cell along specialized membrane structures called intercalated disks. Cardiac muscle is also more responsive to hormonal regulation. For example, cAMP-dependent protein kinases phosphorylate transport proteins and Tn-I, mediating changes in the force of contraction in response to epinephrine.
|
| Smooth muscle can respond to both neural and circulating factors for the stimulus of depolarization. Unlike skeletal muscle, neural input to smooth muscle innervates bundles of smooth muscle cells that cause both phasic (rhythmic) and tonic contractions of the tissue.
|
| page 267 | | | page 268 |
| About 1 in 150 000 patients treated with halothane (gaseous halocarbon) anesthesia or muscle relaxants, responds with excessive skeletal muscle rigidity and severe hyperthermia with a rapid onset, up to 2°C (4°F) within 1 hour. Unless treated rapidly, cardiac abnormalities may be life-threatening; mortality from this condition exceeds 10%. This genetic disease results from excessive or prolonged release of Ca2+ from the SR, most commonly the result of mutations in the Ca2+-release channels within the SR. Excessive release of Ca2+ leads to a prolonged increase in sarcoplasmic Ca2+ concentration. Muscle rigidity results from Ca2+-dependent consumption of ATP, and hyperthermia results from increased metabolism to replenish the ATP. As muscle metabolism becomes anaerobic, lacticacidemia, and acidosis may develop. The cardiac abnormalities result from hyperkalemia, caused by release of potassium ions from muscle; as supplies of ATP are exhausted, muscle is unable to maintain ion gradients across its plasma membrane. Treatment of malignant hyperthermia includes use of muscle relaxants, e.g. dantrolene, an inhibitor of the ryanodine-sensitive Ca2+-channel (Chapter 7), to inhibit Ca2+-release from the SR. Supportive therapy involves cooling, administration of oxygen, correction of blood pH and electrolyte imbalances and also treatment of cardiac abnormalities. |
|