| Structure and nomenclature of gangliosides
|
| The term 'ganglioside' refers to glycolipids that were originally identified in high concentrations in ganglionic cells of the central nervous system. In cells of the nervous system in general, more than 50% of the sialic acid in the cell is present in gangliosides, and the other 50% is in glycoproteins. Gangliosides are also found in the surface membranes of cells of most extraneural tissues, but in these tissues they account for less than 10% of the total sialic acid.
|
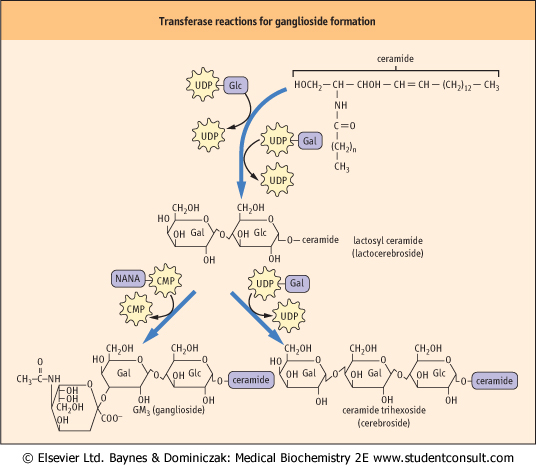
| The nomenclature used to identify the various gangliosides is based on the number of sialic acid residues contained in the molecule, and on the sequence of the carbohydrates (Fig. 26.12). 'GM' means a ganglioside with a single (mono) sialic acid, whereas GD, GT and GQ would indicate two, three and four sialic acid residues in the molecule, respectively. The number after the GM, e.g. GM1 refers to the structure of the oligosaccharide. These numbers were derived from the relative mobility of the glycolipids on thin layer chromatograms; the larger, GM1, gangliosides migrate the most slowly. These complex structures are built up, one sugar residue at a time, in the Golgi apparatus, and are degraded by a series of exoglycosidases in lysosomes (Fig. 26.13). Defects in sequential degradation of glycolipids lead to a number of lysosomal storage diseases, known as cerebrosidoses and gangliosidoses (see box on p. 384).
|
| page 381 |  | | page 382 |
| Figure 26.11 An outline of transferase reactions for elongation of glycolipids and formation of gangliosides. |
| Figure 26.12 Generalized structures of gangliosides. Glc, glucose; Gal, galactose; NANA, N-acetyl neuraminic acid; GalNAc, N-acetylgalactosamine. |
| page 382 | | | page 383 |
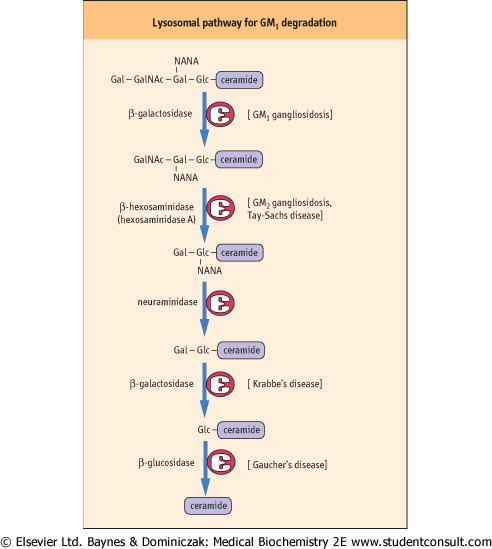
| Figure 26.13 Lysosomal pathway for turnover (degradation) of ganglioside GM1 in human cells. Various enzymes may be missing in specific lipid storage diseases, as indicated in Table 26.1. Gal, galactose; GalNAc, N-acetylgalactosamine; Glc, glucose; NANA, n-acetyl neuraminic acid. |
| LYSOSOMAL STORAGE DISEASES RESULTING FROM DEFECTS IN SPHINGOMYELIN DEGRADATION |
| Niemann-Pick syndrome is a lysosomal storage disease characterized by the formation of lipid-laden phagocytes, known as 'foam cells', that are engorged with sphingomyelin. There is no treatment for this disease, but its inheritance is autosomal recessive and, if carriers are identified, prenatal diagnosis is possible by amniocentesis and assay of sphingomyelinase activity in fetal fibroblasts. |
| Comment. Niemann-Pick syndrome results from a deficiency in the activity of the enzyme, acid sphingomyelinase. In one of the final steps in sphingolipid turnover, this enzyme cleaves the sphingolipid to release choline phosphate and ceramide. Absence of this enzyme leads to accumulation of sphingomyelin in lysosomes. |
| GAUCHER'S DISEASE-A MODEL FOR ENZYME REPLACEMENT THERAPY |
| Gaucher's disease is a lysosomal storage disease in which afflicted individuals are missing the enzyme glucocerebrosidase. This enzyme removes the final sugar from the ceramide, allowing the lipid portion to be further degraded in the lysosomes. This disease is characterized by hepatomegaly and neurodegenerative disease, but there are milder variants that are amenable to treatment by enzyme replacement therapy. |
| For treatment of Gaucher's disease, exogenous β-glucosidase was successfully targeted to the lysosomes of macrophages using a cell surface mannose receptor. In order to do this successfully, it was necessary to produce the recombinant replacement enzyme with N-glycan chains containing terminal mannose residues. This was done by cleaving the glycans of the enzyme produced in mammalian cells with a combination of sialidase (neuraminidase), β-galactosidase and β-hexosaminidase to trim the complex chains down to the mannose core. An alternative recombinant glucosidase has been produced in a baculovirus-infected insect cell system. In this case, the enzyme has a high mannose oligosaccharide that is not processed to complex chains. The recombinant enzymes are administered intravenously. The success in using glucocerebrosidase for treatment of Gaucher's disease has stimulated the development of other lysosomal hydrolases for treatment of lysosomal storage diseases. In a recent study, the lysosomal enzyme, acid lipase, inhibited the progression of atherosclerosis in a hyperlipidemic mouse model. |
| FABRY'S DISEASE - (INCIDENCE 1 IN 100,000) |
| A 30-year-old man is found to have proteinuria at an insurance medical examination. He had been seen over a number of years from around age 10 with headaches, vertigo and shooting pains in his arms and legs. No diagnosis was made and he had grown accustomed to these problems. The physician carefully examined his perineum and scrotum identifying small, raised, red angiokeratoma. |
| Comment. This gentleman has Fabry's disease, which often takes years before a diagnosis is confirmed by measuring α-galactosidase A activity. His insurance was declined as proteinuria is expected to progress to renal failure within 15 years. |
| The principal endothelial depositions of globotriaosylceramide occur in the kidney (leading to proteinuria and renal failure), the heart and brain (leading to myocardial infarction and stroke), and around blood vessels supplying nerves (leading to painful paresthesiae). Recombinant enzyme replacement therapy appears to clear the deposited globotriaosylceramide and initial studies suggest that renal function is maintained. |
| page 383 | | | page 384 |
|
Table 26-1.
Some lipid storage diseases |
| Body_ID: None |
| Lipid storage diseases |
| Body_ID: T026001.50 |
| Disease | Symptoms | Major storage product | Deficient enzymes |
| Body_ID: T026001.100 |
| Tay-Sachs | blindness, mental retardation, death between 2nd and 3rd year | GM2 ganglioside | hexosaminidase A |
| Body_ID: T026001.150 |
| Gaucher's | liver and spleen enlargement, mental retardation in infantile form | glucocerebroside | β-glucosidase |
| Body_ID: T026001.200 |
| Fabry's | skin rash, kidney failure, pain in lower extremities | ceramide trihexoside | α-galactosidase |
| Body_ID: T026001.250 |
| Krabbe's | liver and spleen enlargement, mental retardation | galactocerebroside | β-galactosidase |
| Body_ID: T026001.300 |
| CEREBROSIDOSES AND GANGLIOSIDOSES |
| Tay-Sachs disease is a gangliosidosis in which GM2 accumulates as a result of an absence of hexosaminidase A. Individuals with this disease usually have mental retardation and blindness and die between 2 and 3 years of age. Fabry's disease is a cerebrosidosis resulting from deficiency of α-galactosidase and accumulation of ceramide trihexoside. The symptoms of Fabry's disease are skin rash, kidney failure, and pain in the lower extremities. Patients with this condition benefit from kidney transplants and usually live into early to mid-adulthood. Most of these lysosomal storage diseases appear in several forms (variants), resulting from different mutations in the genome; some variants are more severe and debilitating than others. Although lysosomal storage diseases are relatively rare, they have had a major impact on our understanding of the function and importance of lysosomes. |
| Comment. When cells die, their components, including glycosphingolipids and glycoproteins, are degraded to their individual components. Figure 26.13 presents the pathway for the degradation of a ganglioside such as GM1 in the lysosomes. These organelles contain a number of exoglycosidases and other necessary enzymes that function in a prescribed sequence to degrade complex glycolipids. A number of lysosomal diseases result from the absence of one of these essential glycosidases (Table 26.1). The sphingolipidoses are characterized by lysosomal accumulation of the substrate of the missing enzyme. This either causes lysosomes to lyse and release their hydrolytic enzymes into the cell, or prevents the lysosome from functioning normally. |
| ABO BLOOD-GROUP ANTIGENS ARE GLYCOSPHINGOLIPIDS |
| Blood transfusion replenishes the oxygen-carrying capacity of blood in persons who suffer from blood loss or anemia. The term 'blood transfusion' is something of a misnomer, because it involves only the infusion of washed and preserved red cells. The membranes of the human red blood cells contain a number of blood group antigenic determinants (as many as 100), of which the ABO blood-group system is the best understood and most widely studied. The antigens in this group are derived from a common precursor, called the H substance, and an individual may have type A, type B, type AB, or type O blood. Individuals with type A cells develop natural antibodies in their plasma that are directed against, and will agglutinate type B and type AB red blood cells; whereas those with type B red cells develop antibodies against A substance and will agglutinate type A and type AB blood. Persons with type AB blood have neither A nor B antibodies, and are called 'universal recipients', as they can be transfused with cells of either blood type. Individuals with type O blood have only H substance, not A or B substance, on their red blood cells, and are 'universal donors', as their red blood cells are not agglutinated with either A or B antibodies; they may accept blood only from a type O donor. |
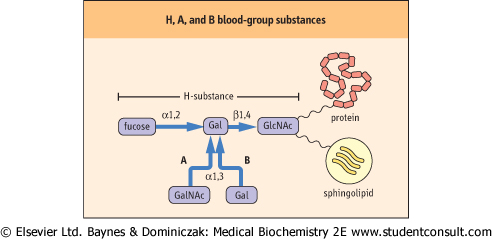
| The ABO blood-group antigens are complex carbohydrates present as components of glycoproteins or glycosphingolipids of red cell membranes. The precursor of the ABO types is the H substance (Fig. 26.14); the H locus codes for a fucosyltransferase. Individuals with type A blood have, in addition to the H substance, an A gene that codes for a specific GalNAc transferase that adds GalNAc α1,3 to the galactose residue of H substance, to form the A-type glycolipid. Individuals with type B blood have a B gene that codes for a galactosyl transferase that adds galactose α1,3 to the galactose residue of H substance, to form the B-type glycolipid. Individuals with type AB blood have both the GalNAc and the galactosyl transferases, and their red blood cells contain both the A and the B substances. Those with type O blood, and only H substance on their red cell glycosphingolipids, do not make either enzyme. Enzymes such as coffee bean α-galactosidase can remove the galactose from type B red cells, an approach that may be tested for increasing the supply of type O red cells. |
| page 384 | | | page 385 |
| OTHER BLOOD GROUP SUBSTANCES ARE ALSO CARBOHYDRATES |
| The Lewis blood group antigens correspond to a set of α1,3(4)-fucosylated glycan structures. The Lewis A antigen (Lea) is synthesized by fucosyltransferase encoded by the Lewis (Le) blood group locus. The Lewis B antigen (Leb) is synthesized by the concerted action of a second fucosyltransferase encoded by the Se (secretor) blood group locus. The nature of the alleles at an individual's Le and Se loci determines the complement of Lewis-active oligosaccharides that that individual will produce. |
| The P blood group antigens are membrane glycosphingolipids on red cells and on other tissues. Again, the glycans in this blood group are synthesized by the sequential action of distinct glycosyltransferases, but so far relatively little is known about the enzymes or genes involved. The physiological function for blood groups is unknown, but the P antigens are associated with the pathophysiology of urinary tract infections and parvovirus infections. The basis for implicating P antigens in urinary tract infections is that uropathogenic strains of E. coli express lectins that bind to the Galα1,4Gal moiety of the Pk and P1 antigens. Clearly more work is necessary to understand the genetics and biochemistry of these and other blood group antigens as well as their roles in physiology and disease. |
| Ganglioside receptor for cholera toxin |
| The galactose-containing cerebrosides and globosides in the plasma membranes of intestinal epithelial cells are binding sites for bacteria. The glycolipids appear to assist in retention of normal intestinal flora (symbionts) in the intestine but, conversely, binding of pathogenic bacteria to these and other glycolipids is believed to facilitate infection of the epithelial cells. The difference between symbiotic and parasitic bacteria depends, in part, on their ability to secrete toxins or to penetrate the host cell after the binding reaction. |
| Intestinal mucosal cells contain ganglioside GM1 (see Fig. 26.12). This ganglioside serves as the receptor to which cholera toxin binds as the first step in its penetration of intestinal cells. Cholera toxin is a hexameric protein secreted by the bacterium Vibrio cholerae. The protein is composed of one A subunit and five B subunits. The protein binds to gangliosides through the B subunits, which enables the A subunit to enter the cell and activate adenylate cyclase on the inner surface of the membrane. The cyclic AMP that is formed then stimulates intestinal cells to export chloride ions, leading to osmotic diarrhea, electrolyte imbalances, and malnutrition. Cholera remains the number one killer of children in the world today. |
| Figure 26.14 Relationship between the H, A, and B blood-group substances. The terminal oligosaccharide is linked through other sugars to proteins and lipids of the red cell membrane. GlcNAc, N-acetylglucosamine; GalNAc, N-acetylgalactosamine; Gal, galactose. |
|