| page 387 |  | | page 388 |
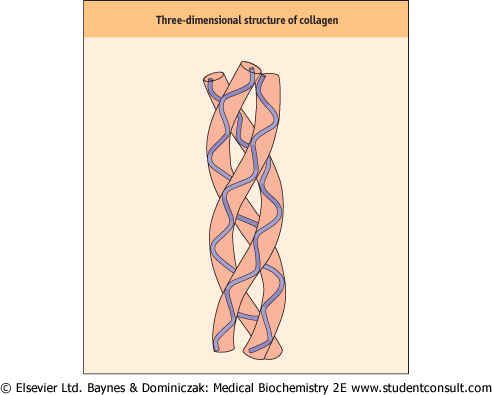
| Figure 27.1 Three-dimensional structure of collagen. Collagen monomer strands assume a left-handed, α-helical tertiary structure. They then associate to form a triple-stranded, right-handed superhelical quarternary structure. |
| Some representative collagens are listed in Table 27.1. The collagen family of proteins can be divided into two main
types: the fibril-forming (fibrillar) and the nonfibrillar collagens.
|
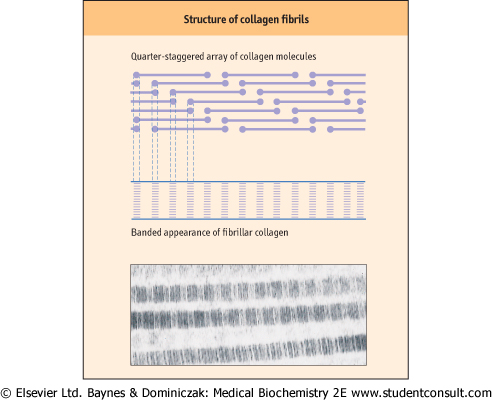
| Figure 27.2 Formation of the quarter-staggered array of collagen molecules in a fibril. The regular overlap of the short, nonhelical termini of the collagen chains yields a regular, banded pattern in the collagen fiber. (Electron micrograph courtesy of Dr Trevor Gray.) |
|
Table 27-1.
Classification and distribution of representative collagens. |
| Body_ID: None |
| Members of the collagen family |
| Body_ID: T027001.50 |
| Type | Class | Distribution |
| Body_ID: T027001.100 |
| I | fibrillar | skin and tendon |
| Body_ID: T027001.150 |
| II | fibrillar cartilage | developing cornea and vitreous humor |
| Body_ID: T027001.200 |
| III | fibrillar | extensible connective tissue, e.g. skin, lung and vascular system |
| Body_ID: T027001.250 |
| IV | network | basement membranes, kidney, vascular wall |
| Body_ID: T027001.300 |
| V | fibrillar | liver, cornea and mucosa |
| Body_ID: T027001.350 |
| VI | beaded filament | most connective tissue |
| Body_ID: T027001.400 |
| IX | FACIT | cartilage, vitreous humor |
| Body_ID: T027001.450 |
| XI | fibril forming | cartilage, bone, placenta |
| Body_ID: T027001.500 |
| XII | FACIT | embryonic tendon and skin |
| Body_ID: T027001.550 |
| XIII | transmembrane domain | widely distributed |
| Body_ID: T027001.600 |
| XIV | FACIT | fetal skin and tendons |
| Body_ID: T027001.650 |
|
| Body_ID: T027001.700 |
FACIT, fibril-associated collagen with interrupted triple helices.
|
Fibril-forming collagens include type I, -II, -III, -V, and -XI collagens (Table 27.1). Collagen fibrils can be formed from a mixture of different fibrillar collagens. Dermal collagen fibrils are hybrids of type I and type III collagen, and fibrils in corneal stroma are hybrids of type I and type IV collagen. Type I is the most abundant fibrillar collagen and occurs in a wide variety of tissues; others have a more limited tissue distribution (see Table 27.1). Type I and related fibrillar collagens form well-organized, banded fibrils and provide high tensile strength to skin, tendons, and ligaments. These collagens are composed of two different α-helical peptide chains, known as α1(I) and α2(I), each containing about 1000 amino acids per chain and having a triple-helical domain structure, [α1(I)]2α2(I), along almost the entire length of the molecule. The collagen fibrils are formed by lateral association of triple helices in a 'quarter-staggered' alignment in which each molecule is displaced by about one-quarter of its length relative to its nearest neighbor (Fig. 27.2); the quarter-staggered
array is responsible for the banded appearance of collagen fibrils in connective tissues. The fibrils are stabilized by both noncovalent forces and interchain crosslinks derived from lysine residues (see below). per chain and having a triple-helical domain structure, [α1(I)]2α2(I), along almost the entire length of the molecule. The collagen fibrils are formed by lateral association of triple helices in a 'quarter-staggered' alignment in which each molecule is displaced by about one-quarter of its length relative to its nearest neighbor (Fig. 27.2); the quarter-staggered
array is responsible for the banded appearance of collagen fibrils in connective tissues. The fibrils are stabilized by both noncovalent forces and interchain crosslinks derived from lysine residues (see below).
|
| page 388 | | | page 389 |
| OSTEOGENESIS IMPERFECTA - (INCIDENCE 1 IN 30 000-50 000) |
| A six-year-old boy was seen in the casualty department with broken tibia and fibula occurring during a soccer game. His 6-foot-tall father explained that he had broken his legs four times while at school. The father's teeth were slightly transparent and discoloured. |
| Comment. Osteogenesis imperfecta (OI), also called brittle-bone disease, is a congenital disease caused by multiple genetic defects in the synthesis of type I collagen. It is characterized by fragile bones, thin skin, abnormal teeth and weak tendons. The majority of the individuals with this disease have mutations in genes encoding α1(I) and α2(I) chains. Many of these mutations are single-base substitutions that convert glycine in the Gly-X-Y repeat to bulky amino acids, preventing the correct folding of the collagen chains into a triple helix and their assembly to form collagen fibrils. |
| The dominance of type 1 collagen in bone explains why bones are predominantly affected. However, there is remarkable clinical variability characterized by bone fragility, osteopenia, variable degrees of short stature, and progressive skeletal deformities. The most common form of OI, with a presentation that is sometimes mistaken for child abuse, has a good prognosis, with fractures decreasing after puberty, though the general reduction in bone mass ensures lifetime risk remains high. Patients frequently develop deafness due to osteosclerosis, partly from recurrent fractures of the stapes. |
| Bisphosphonate drugs, which inhibit osteoclast activity and thereby inhibit normal bone turnover, have reduced the incidence of fractures. Long-term follow-up studies are underway. |
| Nonfibrillar collagens are a heterogeneous group containing triple-helical segments of variable length, interrupted by one or more intervening nonhelical (noncollagenous) segments. This group includes basement membrane collagens (the type IV family), fibril-associated collagens with interrupted triple helices (FACITs), and collagens with multiple triple-helical domains with interruptions, known as multiplexins. Nonfibrillar collagens associate with the fibrillar collagens, forming microfibrils and network or mesh-like structures.
|
| Type IV collagen, with the composition [α1(IV)]2α2(IV), is the major structural component of all basement membranes, where it assembles into a flexible network structure. It contains a long triple-helical domain interrupted by short non-collagenous sequences; these interruptions in the helical domain block continued association of two triple helices, oblige them to find another partner, and thus form the meshwork of interactions. Anomalies in type IV collagen in the glomerular basement membrane (GBM) result in several glomerular diseases including Goodpasture's syndrome (GP). GP is caused by anti-GBM antibodies that specifically binds to type IV collagen of the GBM. Type VI collagen is widely distributed in connective tissues, where it assembles to form beaded filaments or microfibrillar meshwork structures. Each of its three chains contains a short triple-helical domain and large amino (N)-terminal and carboxyl (C)-terminal globular domains. Collagens of the FACIT group have short triple-helical domains interspersed by nonhelical domains. They do not assemble to form fibrils, but associate with the fibrillar collagens.
|
|