| G-protein-coupled receptors
|
G-protein-coupled receptors comprise a superfamily of structurally related receptors for hormones, neurotransmitters, inflammatory mediators, proteinases, taste and odorant molecules, and light photons. A classic example of this class of receptors is the β-adrenergic receptor (for which the ligand is epinephrine ); its structure-function properties have been extensively studied with respect to its activation of signal transduction cascades (see Fig. 38.2). G-protein-coupled receptors are integral membrane proteins characterized by the seven transmembrane-spanning helices within their structure. They generally comprise an extracellular N-terminus, seven transmembrane-spanning α-helices (20-28 hydrophobic amino acids each), three extracellular and intracellular loops, and an intracellular C-terminal tail. Ligands, such as epinephrine, typically bind to the G-protein-coupled receptor by sitting in a pocket formed by the seven transmembrane helices. G-protein-coupled receptors have no intrinsic catalytic domains; they therefore recruit guanine nucleotide-binding proteins (G-proteins), via their third cytoplasmic loop, to couple to their signal transduction elements. ); its structure-function properties have been extensively studied with respect to its activation of signal transduction cascades (see Fig. 38.2). G-protein-coupled receptors are integral membrane proteins characterized by the seven transmembrane-spanning helices within their structure. They generally comprise an extracellular N-terminus, seven transmembrane-spanning α-helices (20-28 hydrophobic amino acids each), three extracellular and intracellular loops, and an intracellular C-terminal tail. Ligands, such as epinephrine, typically bind to the G-protein-coupled receptor by sitting in a pocket formed by the seven transmembrane helices. G-protein-coupled receptors have no intrinsic catalytic domains; they therefore recruit guanine nucleotide-binding proteins (G-proteins), via their third cytoplasmic loop, to couple to their signal transduction elements.
|
| page 543 |  | | page 544 |
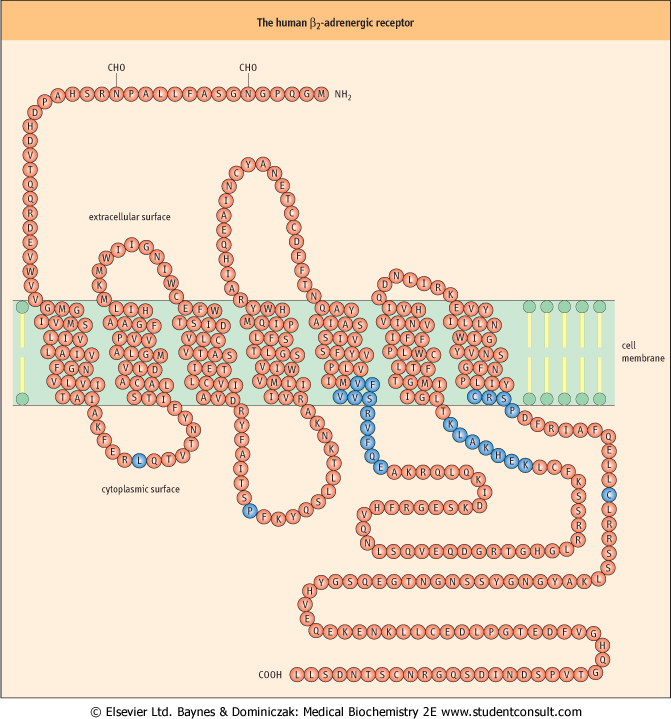
| Figure 38.2 The human β-adrenergic receptor. The sequence of this receptor is organized about the plasma membrane according to the seven membrane-spanning helices model. The ligand-binding site is deep within the plane of the membrane and is formed by amino acids from several of the transmembrane helices. Residues shown in blue in the third cytoplasmic loop and at the beginning of the cytoplasmic tail represent regions that, when deleted or substituted, prevent coupling of the mutated receptor to adenylyl cyclase. The blue residues in the first and second cytoplasmic loops (Leu64 and Pro138) and the carboxyl tail (Cys341) are conserved in many G-protein-coupled receptors and, when mutated, are found to affect markedly both the expression and the functionality of the receptor (see also Chapter 39). |
| page 544 | | | page 545 |
|
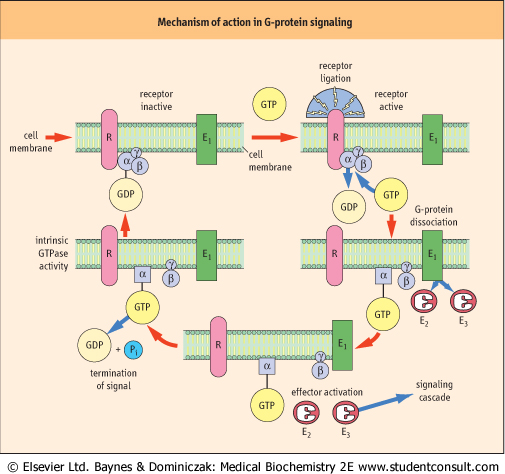
| Figure 38.3 Mechanism of G-protein signaling. In the inactive state, G-proteins exist as heterotrimers with GDP bound tightly to the α-subunit. None of the subunits are integral membrane proteins; however, the G-protein is anchored to the plasma membrane by lipid modification of the γ-subunits (prenylation) and some of the α-subunits (myristoylation of the Giα family). Ligation of the receptor (R) drives exchange of GDP for GTP and induces a conformational change in Gα, which results in a decrease in its affinity both for the receptor and for the βγ-subunits, leading to dissociation of the receptor-G-protein complex. The activated Gα (GTP-bound) or the released βγ-subunits, or both, can then interact with one or more effectors, to generate intracellular second messengers that activate downstream signaling cascades. Signaling is terminated by the intrinsic GTPase activity of the α-subunit, which hydrolyzes GTP to GDP to allow re-association of the inactive, heterotrimeric G-protein, Gαβγ. (See also Fig. 39.4.) |
| BACTERIAL TOXINS WHICH TARGET G-PROTEINS CAUSE A RANGE OF DISEASES |
| A variety of bacterial toxins exert their toxic effects by covalently modifying G-proteins and hence irreversibly modulating their function. For example, cholera toxin (choleragen) from Vibrio cholerae contains an enzyme (subunit A) that catalyzes the transfer of ADP-ribose from intracellular NAD to the α-subunit of Gs; this modification prevents the hydrolysis of Gs-bound GTP, resulting in a constitutively (permanently) active form of the G-protein. The resulting prolonged increase in cAMP concentrations within the intestinal epithelial cells leads to phosphokinase A-mediated phosphorylation of Cl- channels, causing a large efflux of electrolytes and water into the gut, which is responsible for the severe diarrhea that is characteristic of cholera. Enterotoxin action is initiated by specific binding of the B (binding) subunits of choleragen (AB5) to the oligosaccharide moiety of the monosialoganglioside, GM1, on epithelial cells. A similar molecular mechanism has been attributed to the action of the heat-labile enterotoxin, labile toxin, secreted by several strains of Escherichia coli responsible for 'traveler's diarrhea'. |
| In contrast, pertussis toxin (another AB5 toxin) from Bordetella pertussis, the causative agent of whooping cough, catalyzes the ADP-ribosylation of Gi, which prevents Gi from interacting with activated receptors. Hence, the G-protein is inactivated and cannot act to inhibit adenylyl cyclase, activate PLA2 or PLC, open K+ channels, or open and close Ca2+ channels, causing a generalized uncoupling of hormone receptors from their signaling cascades. For example, the α2-adrenergic receptor mediates inhibition of insulin secretion from pancreatic islet cells by inhibiting adenylyl cyclase in a Gi-dependent manner. |
| page 545 | | | page 546 |
| G-proteins constitute a group of regulatory molecules that are involved in the regulation of a diverse range of biological processes, including signal transduction, protein synthesis,
intracellular trafficking (targeted delivery to the plasma membrane or intracellular organelles) and exocytosis, as well as cell movement, growth, proliferation and differentiation. The G-protein superfamily predominantly comprises two major subfamilies: the small, monomeric Ras-like G-proteins (see Chapter 41) and the heterotrimeric G-proteins. Heterotrimeric G-proteins regulate the transduction of transmembrane signals from cell-surface receptors to a variety of intracellular effectors, such as adenylyl cyclase, phospholipase C (PLC), cGMP-phosphodiesterase (PDE) and ion-channel effector systems (Fig. 38.3). These G-proteins consist of three distinct classes of subunits: α (39-46 kDa), β(37 kDa), and γ (8 kDa). In general, effector specificity is conferred by the α-subunit, which contains the GTP-binding site and an intrinsic GTPase activity. However, it is now widely accepted that βγ complexes can also directly regulate effectors such as phospholipase A2 (PLA2), PLC-β isoforms, adenylyl cyclase and ion channels in mammalian systems, as well as cellular responses such as mating-factor receptor pathways in yeast. Four major subfamilies of α-subunit genes have been identified on the basis of their cDNA homology and function: Gs, Gi, Gq, and G12 (Table 38.2). Many of these Gα subunits have been shown to exhibit a rather ubiquitous pattern of expression in mammalian systems, at least at the mRNA level, but it is also clear that certain α-subunits have a tissue-restricted profile of expression. Moreover, there is evidence of differential expression of α-subunits during cellular development.
|
| G-proteins act as molecular switches
|
| Heterotrimeric G-proteins regulate transmembrane signals by acting as a molecular switch, linking cell-surface G-protein-coupled receptors to one or more downstream signaling molecules (see Fig. 38.3).
|
| Ligation of the receptor initiates an interaction with the inactive, GDP-bound heterotrimeric G-protein. This interaction drives exchange of GDP for GTP, inducing a conformational change in Gα, which results in a decrease in its affinity both for the receptor and for the βγ-subunits, leading to dissociation of the receptor-G-protein complex. The activated Gα (GTP-bound) or released βγ-subunits, or both, can then interact with one or more effectors to generate intracellular second messengers, which activate downstream signaling cascades. Signaling is terminated by the intrinsic GTPase activity of the α-subunit, which hydrolyses GTP to GDP to allow reassociation of the inactive heterotrimeric G-protein (Gαβγ).
|
|