| ANALYSIS OF PROTEIN STRUCTURE
|
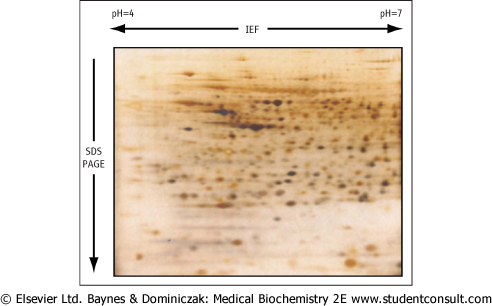
| A proteome is defined as the full complement of proteins produced by a particular genome. Proteomics is an important field of study of protein properties and is defined as the qualitative and quantitative comparison of proteomes under different conditions with the goal of further unraveling biological process. To analyze the proteome of a cell, proteins from a cell are extracted and subjected to 2-dimensional gel electrophoresis. Individual protein spots are extracted and digested with proteases. Small peptides from such a gel are sequenced by mass spectrometry, permitting the identification of the protein. A typical analysis of a rat liver extract is shown in Fig. 2.16. |
| Figure 2.16 Two-dimensional gel electrophoresis. A crude rat liver extract was first subjected to isoelectric focusing (IEF) in cylindrical gel within the pH range 4-7. The gel was then laid horizontally on a second slab gel and separated by SDS-PAGE according to molecular mass. The gel was then stained with Coomassie Blue. |
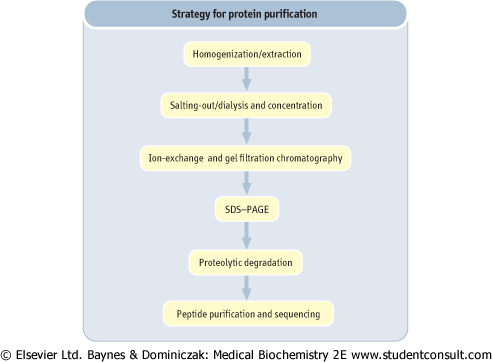
The typical steps in the purification of a protein are summarized in Figure 2.17. Once purified, for the determination of its amino acid composition, a protein is subjected to hydrolysis, commonly in 6 mol/L HCl at 110°C in a sealed and evacuated tube for 24-48 h. Under these conditions, tryptophan, cysteine, and most of the cystine are destroyed, and glutamine and asparagine are quantitatively deaminated to give glutamate and aspartate, respectively. Recovery of serine and threonine is incomplete and decreases with increasing time of hydrolysis. Alternative hydrolysis procedures may be used for measurement of tryptophan, while cysteine and
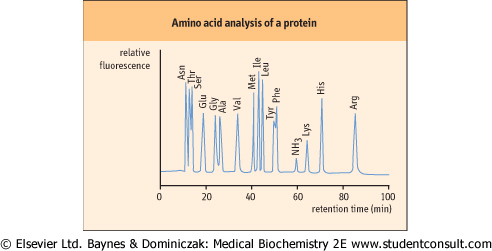
cystine may be converted to an acid-stable cysteic acid prior to hydrolysis. Following hydrolysis, the free amino acids are separated on an automated amino acid analyzer using an ion-exchange column, or by reversed-phase high-performance liquid chromatography (HPLC). The amino acids are reacted with chromogenic or fluorogenic reagents, such as ninhydrin or dansyl chloride, Edman's reagent (see below), or o-phthalaldehyde. These techniques allow the measurement of as little as 1 pmol of each amino acid. A typical elution pattern of amino acids in a purified protein is shown in Figure 2.18. are separated on an automated amino acid analyzer using an ion-exchange column, or by reversed-phase high-performance liquid chromatography (HPLC). The amino acids are reacted with chromogenic or fluorogenic reagents, such as ninhydrin or dansyl chloride, Edman's reagent (see below), or o-phthalaldehyde. These techniques allow the measurement of as little as 1 pmol of each amino acid. A typical elution pattern of amino acids in a purified protein is shown in Figure 2.18.
|
| page 21 |  | | page 22 |
| Figure 2.17 Strategy for protein purification. Purification of a protein involves a sequence of steps in which contaminating proteins are removed, based on difference in size, charge and hydrophobicity. Purification is monitored by SDS-PAGE. The primary sequence of the protein is determined by automated Edman degradation of peptides. The three-dimensional structure of the protein may be determined by X-ray crystallography. |
| Figure 2.18 Typical chromatogram from an amino acid analysis by cation-exchange chromatography. A protein hydrolysate is applied to the cation exchange column in a dilute buffer at acidic pH (∼3.0), at which all amino acids are positively charged. The amino acids are then eluted by a gradient of increasing pH and salt concentrations. The most anionic (acidic) amino acids elute first, followed by the neutral and basic amino acids. Amino acids are derived by post-column reaction with a fluorogenic compound, such as o-phthalaldehyde. |
| Determination of the primary structure of proteins
|
Information on protein primary sequence of a protein is essential for understanding its functional properties, the identification of the family to which the protein belongs, as well as characterization of mutant proteins that cause disease. A protein may be cleaved first by digestion by specific endoproteases, such as trypsin (see Chapter 5), V8 protease, or lysyl endopeptidase, to obtain peptide fragments. Trypsin cleaves peptide bonds on the C-terminal side of arginine and lysine residues, provided the next residue is not proline. Lysyl endopeptidase is also frequently used to cleave at the C-terminal side of lysine. Cleavage by chemical reagents such as cyanogen bromide is also useful. Cyanogen bromide cleaves on the C-terminal side of methionine residues. Before cleavage, proteins with cysteine and cystine residues are reduced by 2-mercaptoethanol (MeSH) and then treated with iodoacetate to form carboxymethylcysteine residues. This avoids spontaneous formation of inter- or intra-molecular disulfides during analyses.
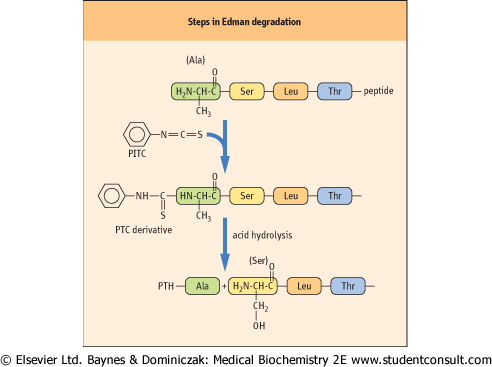
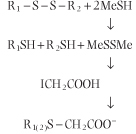 The cleaved peptides are then subjected to reversed-phase HPLC to purify the peptide fragments, and then sequenced on an automated protein sequencer, using the Edman degradation technique (Fig. 2.19). The sequence of overlapping peptides is then used to obtain the primary structure of the protein. Mass spectrometry may also be used to obtain both the molecular mass and sequence of polypeptides simultaneously (see below). Both techniques can be applied directly to proteins or peptides recovered from SDS-PAGE or two-dimensional electrophoresis (IEF plus SDS-PAGE). Once the partial amino acid sequence is obtained, one can determine the nucleotide sequence of the DNA that encodes this polypeptide segment. After chemically synthesizing this DNA, it can be used to identify and isolate the gene containing its nucleotide sequence (Chapter 34).
The cleaved peptides are then subjected to reversed-phase HPLC to purify the peptide fragments, and then sequenced on an automated protein sequencer, using the Edman degradation technique (Fig. 2.19). The sequence of overlapping peptides is then used to obtain the primary structure of the protein. Mass spectrometry may also be used to obtain both the molecular mass and sequence of polypeptides simultaneously (see below). Both techniques can be applied directly to proteins or peptides recovered from SDS-PAGE or two-dimensional electrophoresis (IEF plus SDS-PAGE). Once the partial amino acid sequence is obtained, one can determine the nucleotide sequence of the DNA that encodes this polypeptide segment. After chemically synthesizing this DNA, it can be used to identify and isolate the gene containing its nucleotide sequence (Chapter 34).
|
| Determination of the three-dimensional structure of proteins
|
| X-ray crystallography and nmR spectroscopy are usually used for the determination of the three-dimensional structure of proteins.
|
| page 22 | | | page 23 |
| Figure 2.19 Steps in Edman degradation. The Edman degradation method sequentially removes one residue at a time from the amino end of a peptide. Phenyl isothiocyanate (PITC) converts the N-terminal amino group of the immobilized peptide to a phenylthiocarbamyl derivative (PTC amino acid) in alkaline solution. Acid treatment removes the first amino acid as the phenylthiohydantoin (PTH) derivative, which is identified by HPLC. |
| X-ray crystallography involves the diffraction of X-rays by the electrons of the atoms constituting the molecule. However, since the X-ray diffraction caused by an individual molecule is immeasurably weak, the protein must exist in the form of a well-ordered crystal, in which each molecule has the same conformation in a specific position and orientation on a three dimensional lattice. Based on diffraction of a collimated beam of electrons, the distribution of the electron density, and thus the location of atoms, in the crystal can be calculated to determine the structure of the protein. For protein crystallization, the most frequently used method is the hanging drop method which involves the use of a simple apparatus that permits a small portion of a protein solution (typically 10 μl droplet containing 0.5-1 mg/protein) to evaporate gradually to reach the saturating point at which the protein begins to crystallize. nmR spectroscopy is usually used for structural analysis of small organic compounds, but high-field nmR is also useful for determination of the structure of a protein in solution and complements information obtained by X-ray crystallography. Kurt Wuthrich was awarded the 2002 Nobel Prize in chemistry for his applications of nmR to macromolecules in solution.
|
|