| Hemostasis means 'the arrest of bleeding'
|
| After tissue injury that ruptures smaller vessels (including everyday trauma, injections, surgical incisions, and tooth extractions), a series of interactions between the vessel wall and the circulating blood normally occur, resulting in cessation of blood loss from injured vessels within a few minutes (hemostasis). Hemostasis results from effective sealing of the ruptured vessels by a hemostatic plug composed of blood platelets and fibrin. Fibrin is derived from circulating fibrinogen, whereas platelets are small cell fragments that circulate in the blood and have an important role in the initiation of hemostasis.
|
| Hemostasis requires the effective, coordinated function of blood vessels, platelets, coagulation factors and the fibrinolytic system
|
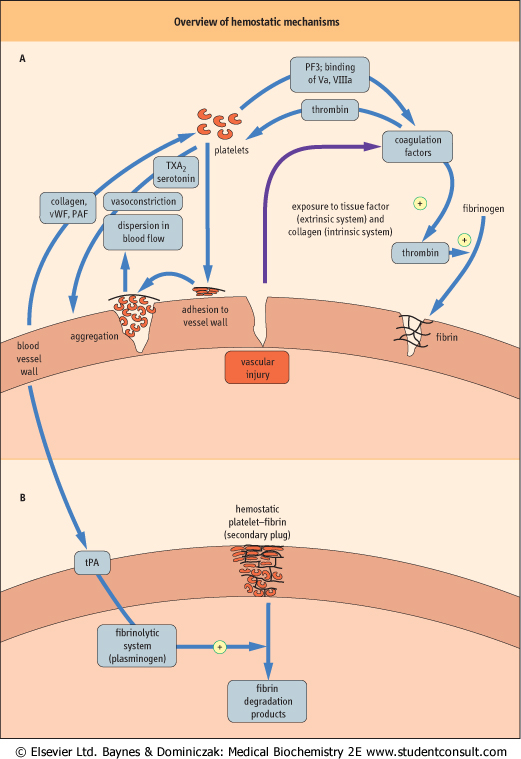
| Figure 6.1 provides an overview of hemostatic mechanisms, and illustrates some of the interactions between blood vessels, platelets, and the coagulation system in hemostasis; each of these components of hemostasis also interacts with the fibrinolytic system. The initial response of small blood vessels to injury is arteriolar vasoconstriction, which temporarily reduces local blood flow. Flow reduction transiently reduces blood loss, and may also promote formation of the platelet-fibrin plug. Activation of blood platelets is followed by their adhesion to the vessel wall at the site of injury, and their subsequent aggregation to each other, building up an occlusive platelet mass that forms the initial (primary) hemostatic plug. This platelet plug is friable and, unless subsequently stabilized by fibrin, will be washed away by local blood pressure when vasoconstriction reverses.
|
| page 63 |  | | page 64 |
| Figure 6.1 Overview of hemostatic mechanisms. (A) Vascular injury sets in motion a series of events that culminate in formation of a primary plug of platelets. This can be dispersed by blood flowing through the vessel unless the plug is stabilized. (B) The primary plug is stabilized by a network of fibrin (formed from crosslinked fibrinogen). The secondary plug is stable and is degraded only when the fibrinolytic system has been activated. PAF, platelet activating factor; PF3, platelet factor 3; tPA, tissue-type plasminogen activator; TXA2, thromboxane A2; Va, activated coagulation factor V; VIIIa, activated coagulation factor VIII; vWF, von Willebrand factor. |
| page 64 | | | page 65 |
Vascular injury also activates coagulation factors, which interact sequentially to form thrombin , which converts circulating soluble plasma fibrinogen to insoluble, crosslinked fibrin. This forms the subsequent (secondary) hemostatic plug, which is relatively resistant to dispersal by blood flow
or fibrinolysis. There are two pathways of the activation of coagulation factors: the extrinsic pathway which is initiated by the exposure of the flowing blood to tissue factor, released from subendothelial tissue; and the intrinsic pathway which has an important amplification role in generating thrombin and fibrin. , which converts circulating soluble plasma fibrinogen to insoluble, crosslinked fibrin. This forms the subsequent (secondary) hemostatic plug, which is relatively resistant to dispersal by blood flow
or fibrinolysis. There are two pathways of the activation of coagulation factors: the extrinsic pathway which is initiated by the exposure of the flowing blood to tissue factor, released from subendothelial tissue; and the intrinsic pathway which has an important amplification role in generating thrombin and fibrin.
|
| The lysis of fibrin is equally important to health as is its formation
|
| Hemostasis is a continuous process throughout life, and would result in excessive fibrin formation and vascular occlusion if unchecked. Evolution has therefore produced a fibrinolytic system; this is activated by local fibrin formation, resulting in local generation of plasmin, an enzyme which digests fibrin plugs (in parallel with tissue repair processes), thus maintaining vascular patency. Digestion of fibrin results in generation of circulating fibrin degradation products (FDP). These are detectable in plasma of healthy individuals at low concentration, which illustrates that fibrin formation and lysis are continuing processes in health.
|
| Excessive bleeding may result from defects in each of the components of hemostasis, which may be caused by disease (congenital or acquired) or by antithrombotic drugs (Table 6.1).
|
| The vascular, platelet, coagulation and fibrinolytic components of hemostasis will now be discussed in turn.
|
|
Table 6-1.
Congenital and acquired causes of excessive bleeding. |
| Body_ID: None |
| Causes of excessive bleeding |
| Body_ID: T006001.50 |
| | Congenital | Acquired |
| Body_ID: T006001.100 |
| vessel wall | disorders of collagen synthesis (Ehlers-Danlos syndrome) | vitamin C deficiency (scurvy) corticosteroid excess |
| Body_ID: T006001.150 |
| platelets | vWF deficiency (von Willebrand disease) platelet GPIb-IX deficiency (Bernard-Soulier syndrome) platelet GPIIb-IIIa deficiency (Glanzmann's thrombasthenia) | antiplatelet drugs (e.g. aspirin) defective formation of platelets excessive destruction of platelets |
| Body_ID: T006001.200 |
| coagulation | coagulation factor deficiencies (hemophilias)-factor VIII factor IX factor XI fibrinogen etc. | vitamin K deficiency (factors II, VII, IX, X) oral anticoagulants (vitamin K antagonists, e.g. warfarin) liver disease disseminated intravascular coagulation (DIC) |
| Body_ID: T006001.250 |
| fibrinolysis | antiplasmin deficiency PAI-1 deficiency | fibrinolytic drugs (e.g. tPA, urokinase, streptokinase) |
| Body_ID: T006001.300 |
|
| Body_ID: T006001.350 |
GPIb-IX, GPIIb-IIIa, glycoprotein receptors Ib-IX and IIb-IIIa; PAI-1, plasminogen activator inhibitor type 1.
|
|