| Vascular injury has a key role in initiating local formation of the platelet-fibrin plug and in its subsequent removal by the fibrinolytic system
|
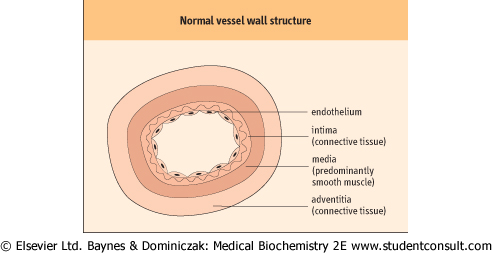
| All blood vessels are lined by a flat sheet of endothelial cells, which have important roles in the interchange of chemicals, cells, and microbes between the blood and the body tissues. Endothelial cells in the smallest blood vessels (capillaries) are supported by a thin layer of connective tissue, rich in collagen fibers, called the intima. In veins, a thin layer (the media) of contractile smooth muscle cells allows some venoconstriction: for example, superficial veins under the skin constrict in response to surface cooling. In arteries and arterioles, a well-developed muscle layer allows powerful vasoconstriction, including the vasoconstriction after local injury that forms part of the hemostatic response. Larger vessels also have a supportive connective tissue outer layer (the adventitia). Figure 6.2 illustrates the structure of the vessel wall.
|
| Normal endothelium has an antithrombotic surface
|
Intact normal endothelium does not initiate or support platelet adhesion or blood coagulation. Its surface is antithrombotic. This thrombo-resistance is partly due to endothelial production of two potent vasodilators and inhibitors of platelet function: prostacyclin (prostaglandin I2, PGI2) and nitric oxide , otherwise known as endothelium-derived relaxing factor (EDRF) (see box on p. 66). , otherwise known as endothelium-derived relaxing factor (EDRF) (see box on p. 66).
|
| Figure 6.2 Normal vessel wall structure. |
| page 65 |  | | page 66 |
| PROSTACYCLIN AND NITRIC OXIDE ARE GENERATED IN THE VESSEL WALL |
| Biochemical mediators of vasoconstriction and vasodilatation |
| The diameters of arteries and arterioles throughout the body continuously alter to regulate blood flow according to local and general metabolic and cardiovascular requirements. Control mechanisms include neurogenic (sympathetic/adrenergic; see Chapter 39) and myogenic pathways, and local biochemical mediators, including prostacyclin (PGI2) and nitric oxide. |
| Prostacyclin is the major arachidonic acid metabolite formed by vascular cells. It is a potent vasodilator, and also a potent inhibitor of platelet aggregation. It has a short half-life in plasma (3 minutes). |
| Nitric oxide is also a potent vasodilator formed by vascular endothelial cells, also with a short half-life. It was initially termed endothelium-derived relaxing factor (EDRF). In common with that of prostacyclin, its generation by endothelial cells is enhanced by many compounds, and also by blood flow and shear stress. In the normal circulation, nitric oxide appears to have a key role in flow-mediated vasodilatation. It is synthesized by two distinct forms of endothelial nitric oxide synthase (eNOS): constitutive and inducible. Constitutive eNOS rapidly provides relatively small amounts of nitric oxide for short periods, related to vascular flow regulation. The beneficial effects of nitrate drugs in hypertension and angina may partly reflect their effects on this pathway (see Chapter 17). Inducible eNOS is stimulated by cytokines in inflammatory reactions, and releases large amounts of nitric oxide for long periods. Its suppression by glucocorticoids may partly account for their anti-inflammatory effects. |
| Both prostacyclin and nitric oxide appear to exert their vasodilator actions by diffusing locally from endothelial cells to vascular smooth muscle cells (see Fig. 6.2), where they stimulate guanylate cyclase, resulting in increased formation of cyclic guanosine 3',5'-monophosphate (cGMP) and relaxation of vascular smooth muscle, probably via alteration of the intracellular calcium concentration (see Chapter 38). |
| The vasoconstriction that occurs after vascular injury is partly mediated by two platelet activation products: serotonin (5-hydroxytryptamine), and thromboxane A2 (TXA2; see Fig. 6.1), a product of platelet prostaglandin metabolism. In
addition, after a vascular injury that disrupts the endothelial cell lining, flowing blood is exposed to subendothelial collagen, which activates the intrinsic pathway of blood coagulation. The endothelial cell damage also exposes flowing blood to subendothelial tissue factor, which activates the extrinsic pathway of blood coagulation (see Fig. 6.1).
|
| PLATELETS PRODUCE THROMBOXANE A2 |
| It has already been noted that PGI2, the major arachidonic acid metabolite formed by vascular cells is a potent vasodilator and inhibitor of platelet aggregation. In contrast, the major arachidonic acid metabolite formed by platelets is TXA2, which is a potent vasoconstrictor and stimulates platelet aggregation. In common with prostacyclin, TXA2 has a short half-life. In the late 1970s, Salvador Moncada and John Vane contrasted the effects of PGI2 and TXA2 on blood vessels and platelets, and hypothesized that a balance between these two compounds was important in the regulation of hemostasis and thrombosis. |
| Congenital deficiencies of cyclo-oxygenase or thromboxane synthase (the enzymes involved in TXA2 synthesis) result in a mild bleeding tendency. Ingestion of even low doses of acetylsalicylic acid (aspirin) irreversibly acetylates cyclo-oxygenase and suppresses TXA2 synthesis and platelet aggregation for several days, resulting in an antithrombotic effect and a mild bleeding tendency. Bleeding is especially likely from the stomach, as a result of the formation of stomach ulcers secondary to the inhibition of cytoprotective gastric mucosal prostaglandins by aspirin. Although in persons at high risk of arterial thrombosis (e.g. previous myocardial infarction or stroke) this bleeding tendency is outweighed by a reduction in risk of thrombosis, aspirin is contraindicated in individuals with a history of bleeding disorders, or existing stomach or duodenal ulcers. |
| Exposure of flowing blood to collagen as a result of endothelial damage also stimulates platelet activation. Platelets bind to collagen via von Willebrand factor (vWF), which is released from the endothelial cells. vWF in turn binds both to collagen fibers and to platelets (via a platelet
membrane glycoprotein receptor, GPIb-IX). Platelet activating factor (PAF) from the vessel wall may also activate platelets in hemostasis (see Fig. 6.1).
|
| Collagen has a key role in the structure and the hemostatic function of small blood vessels
|
| Because collagen has a key role in the structure and the hemostatic function of small blood vessels, vascular causes of excessive bleeding include congenital or acquired deficiencies of collagen synthesis (see Table 6.1). Congenital disorders include the rare Ehlers-Danlos syndrome. Acquired disorders include the relatively common vitamin C deficiency, scurvy (see Chapter 10), and excessive exogenous or endogenous corticosteroids.
|
|