| Normally arterial endothelium repels cells and inhibits blood clotting
|
| The lumen of a healthy artery is lined by a confluent layer of endothelial cells. Normal endothelium forms a surface which repels cells floating in plasma (including platelets), and which is strongly antithrombotic. The arterial wall consists three well-defined layers: the subendothelial layer is called the intima, the middle one, media (this one contains vascular smooth muscle cells, VSMC), and the outer one is adventitia, composed of looser connective tissue and containing relevant nerves.
|
| Substances can penetrate endothelium either through junctions between the endothelial cells, or by transgressing the cells themselves. An important function, controlled by the endothelium, is the ability of blood vessels to dilate (vasodilatation) and to constrict (vasoconstriction) and thus regulate tissue and organ blood flow.
|
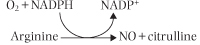
The endothelium controls vasodilatation by secreting the endothelium-derived relaxing factor (EDRF), which is a gas, nitric oxide (NO) (NO)
|
Nitric oxide is synthesized from l-arginine by endothelial NO synthase (eNOS).
|
| MEASUREMENT OF C-REACTIVE PROTEIN MAY REFLECT INFLAMMATORY PHENOMENA ASSOCIATED WITH ATHEROGENESIS |
| It has been long known that the inflammatory reaction associated with infection or trauma can be detected by measuring the concentration of plasma C-reactive protein (CRP), a protein produced in the liver in response to stimulation by pro-inflammatory cytokines. Its name comes from its binding to the capsular polysaccharide of bacteria such as S. pneumoniae by which way it mediates their clearance. However, measurements of small increases in CRP concentration which require a special, highly sensitive analytical method, demonstrated the association of such and may identify persons at risk who might have normal lipid concentrations. Increased plasma concentrations of other pro-inflammatory molecules such as interleukin-6 (IL-6) and serum amyloid A, have also been associated with coronary heart disease. |
| The activity of eNOS is controlled by the intracellular calcium concentration. There are two isoenzymes of NOS: eNOS that is constitutively (constantly) expressed in the endothelium, and inducible (iNOS) found in VSMC and in macrophages. Nitric oxide causes vasodilatation via stimulation of guanylate cyclase and cyclic GMP production (see Chapter 38).
|
| Current data suggest that low levels of NO contribute to development of high blood pressure (hypertension). Also, drugs such as glyceryl trinitrate, which relieve chest pain caused by inadequate blood supply to the heart muscle (angina pectoris) by dilating coronary arteries, act by releasing NO.
|
| Atherosclerosis is a disease of the cardiovascular system, affecting the vessel wall and leading to the narrowing of arteries, or to their complete blockage
|
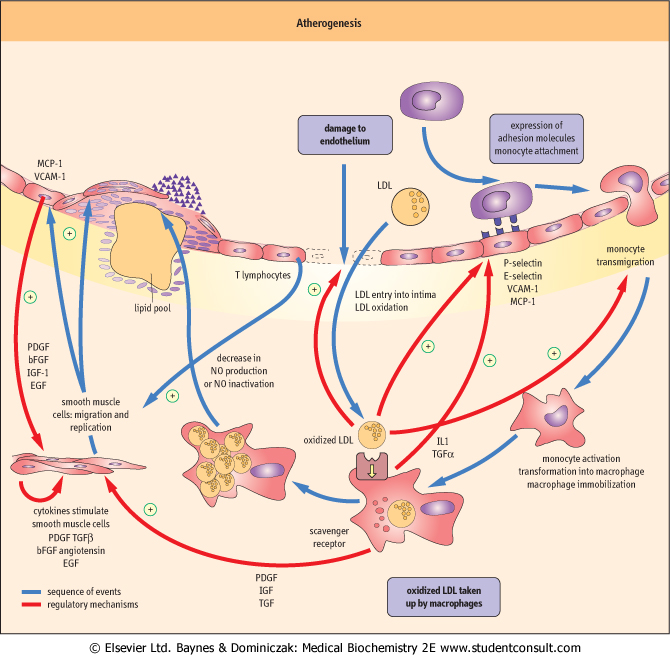
| Clinically this may cause myocardial infarction (resulting from a complete blockage of coronary artery supplying the heart), stroke (a blockage of an artery supplying the brain), or peripheral vascular disease (a condition where narrowing of leg arteries leads to a characteristic pain on walking, known as intermittent claudication). Atherosclerosis is a complex process. Its main components are endothelial dysfunction, lipid deposition, and inflammatory reaction in the vascular wall (Fig. 17.8): all three eventually result, not only in the formation of atherosclerotic plaques, but in the remodeling of the entire arterial wall. The whole process is set in motion, and is maintained, by a intense cross-talk between endothelial cells, VSMC, and plasma-derived inflammatory cells, macrophages, and lymphocytes (Fig. 17.9). The cross-talk involves an array of chemokines, cytokines, and growth factors (see Chapter 37), which cause attraction of cells to the sites of atherosclerotic lesions, induce cell migration, proliferation, apoptosis, and the excess production of extracellular matrix.
|
| The development of atherosclerosis is initiated by endothelial dysfunction
|
| The key early event in atherosclerosis is the damage to the endothelium (Fig. 17.8). This may be caused by excess of lipoproteins, hypertension, diabetes, or the components of cigarette smoke. Initially the damage is functional only. The endothelium becomes more permeable to lipoproteins which move beneath the endothelial layer, in the underlying intima. Also, the endothelium loses its cell-repellent quality, and allows inflammatory cells into the vascular wall. Later, the endothelium may become physically damaged, or even completely destroyed.
|
| page 236 |  | | page 237 |
|
| Figure 17.8 Atherogenesis: the process. Atherogenesis is driven by signals mediated by cytokines and growth factors generated by all the major types of cells participating in the process: endothelial cells, macrophages, T lymphocytes and vascular smooth muscle cells (VSMC). There are multiple activation paths: for instance, the expression of MCP-1 and VCAM-1 may be stimulated by signals generated macrophages as well as by the oxidized LDL. VSMC may be stimulated by the dysfunctional endothelial cells, by macrophages, and by T lymphocytes (note also the autocrine activation). Note that a hormone, angiotensin II also participates in these processes. MCP-1: monocyte chemoattractant protein 1, VCAM-1: vascular cell adhesion molecule 1, ICAM-1: intracellular cell adhesion molecule 1, TNFβ: tumor necrosis factor beta, TNFα: tumor necrosis factor alpha, IFNγ: interferon gamma, NO: nitric oxide, PDGF: platelet-derived growth factor, bFGF: basic fibroblast growth factor, IGF-1: insulin-like growth factor 1, EGF: epidermal growth factor, TGFβ: transforming growth factor beta, IL-1: interleukin 1. |
| Dysfunctional endothelium expresses the so-called adhesion molecules on its surface. Molecules called selectins mediate the initial, 'rolling' interaction of cells with the endothelium. The key molecule that promotes adhesion of monocytes (white blood cells that are the precursors of macrophages) and T lymphocytes is the vascular cell-adhesion molecule-1 (VCAM-1) (see also Fig. 25.14). The adhering cells are then stimulated by the monocyte chemoattractant protein-1 (MCP-1) to cross the endothelium and lodge in the intima.
|
| page 237 | | | page 238 |
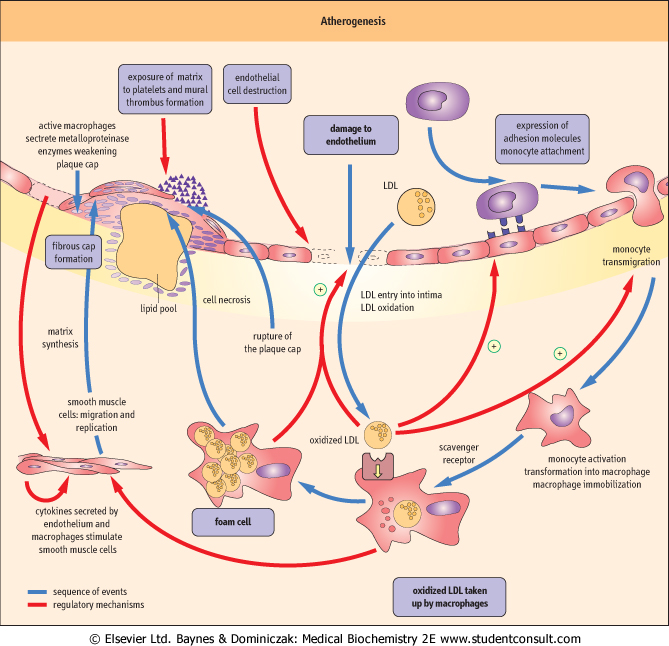
| Figure 17.9 Atherogenesis: role of growth factors and cytokines. Atherogenesis involves endothelial dysfunction, arterial deposition of lipids, inflammatory reaction, and the migration and proliferation of the arterial smooth muscle cells. Note the key role of lipid oxidation in the formation of lipid-laden cells and the lipid center of the atherosclerotic plaque. The sequence of events, and their control by cytokines and growth factors are described in the text. (Compare Fig. 17.8.) |
| In the intima, under the influence of monocyte colony-stimulating factor (M-CSF) secreted by the endothelial cells and VSMC, the monocytes transform into resident macrophages. T lymphocytes also enter the vascular wall at this stage. Macrophages produce reactive oxygen species, which oxidize LDL present in the intima. The expression of macrophage scavenger receptors (such as the scavenger receptor A, and CD36) also increases.
|
| The hormone angiotensin II (see Chapter 22) also contributes to cell entry into intima by contributing to the increased expression of VCAM-1 and MCP-1. In addition, the production of nitric oxide decreases in the damaged endothelium. NO normally reduces monocyte adhesion and VSMC migration and proliferation. Its decrease also shifts the balance between vascular vasodilatation and vasoconstriction towards the latter.
|
| page 238 | | | page 239 |
| Lipid entry into the arterial wall is a key process in atherogenesis
|
| Another factor which contributes to the induction of VCAM-1 and MCP-1 is hypercholesterolemia. However, it is the physical entry of lipids into the vascular wall, which is another hallmark of atherosclerosis. The lipoproteins of smaller size, the remnants and the LDL, are the most atherogenic partly because they enter the vascular wall more easily. Moreover, after leaving plasma, the LDL are modified in a way which enhances their potential to cause damage: while in plasma they are protected against oxidation by alpha-tocopherol (vitamin E), and ubiquinol, and by plasma antioxidants such as vitamin C and β-carotene. However, that protection does not apply to the extracellular space where the concentration of antioxidants is much lower. Therefore, once LDL exit plasma, their phospholipids and fatty acids oxidize. Enzymes such as lipoxygenases, myeloperoxidase, and NADPH oxidases present in the activated macrophages facilitate LDL oxidation.
|
| Oxidized LDL become yet another factor which stimulates the expression of VCAM-1 and MCP-1. They are also cytotoxic to the endothelial cells and are mitogenic for macrophages. Most importantly though, the LDL 'driver' apolipoprotein, apoB100, once oxidized, binds to the scavenger receptors, and these receptors are not subject to regulation by the intracellular cholesterol level. Therefore, macrophages which take up oxidized LDL overload themselves with lipids. As a result, they change into the so-called foam cells: conglomerates of these cells are visible in the arterial walls as yellow patches, and are called fatty streaks. Dying foam cells release lipid that pools within the intima. These lipid accumulations become centers of the atherosclerotic plaques.
|
| Migration and proliferation of the vascular smooth muscle cells change the structure of the vascular wall
|
| All the above is accompanied by profound changes in the behavior of VSMC. VSMC stimulated by growth factors such as the platelet-derived growth factor (PDGF), the epidermal growth factor (EGF) and the insulin-like growth factor-1 (IGF-1), proliferate and migrate toward the lumen of the arterial wall (see also Chapters 38 and 41). They also secrete adhesion molecules and MCP-1 and, as do the endothelial cells, a range of cytokines and growth factors such as interleukin-1 (IL-1), and tumor necrosis factor (TNF-α). Importantly, activated VSMC also synthesize extracellular matrix, in particular collagen, which is deposited in the forming plaque. As a result, the normally ordered structure of the arterial wall becomes completely disrupted, and the forming plaque may protrude into the lumen of the artery, interfering with the flow of blood.
|
| Inflammation plays a fundamental role in atherogenesis
|
| The exit of monocytes and T leukocytes from plasma and their activation in the intima are parts of the inflammatory response. Normally, such response is initiated by an antigen or trauma. However, no specific antigen capable of intiating atherogenesis has been identified to date.
|
| It is possible that there is a molecular mimicry between the antigens involved in atherosclerosis and the exogenous pathogens (Chapter 36). Such putative antigen(s) might be infectious agents, or molecules modified by reactive oxygen species. For instance, phosphorylcholine group found in the oxidized LDL is also a component of the capsular polysaccharide of bacteria. Indeed, oxidized LDL remains a candidate antigen which could be responsible for the stimulation of inflammatory reaction in atherosclerosis.
|
| Atherogenesis involves both innate and adaptive immunity (see Chapter 36). Innate immunity includes recognition by scavenger receptors A, CD36 (Chapter 36). When molecules which possess patterns encoded in immune memory bind to these receptors, they activate cells through, for instance, the pathway involving the transcription factor NF-κB. We also know that T cells, involved in adaptive immunity, are present in both early and late atherosclerotic lesions. Finally, circulating IgG and IgM-type antibodies against modified LDL have been identified in plasma.
|
| Atherosclerotic plaques grow slowly, but the real danger is their sudden rupture
|
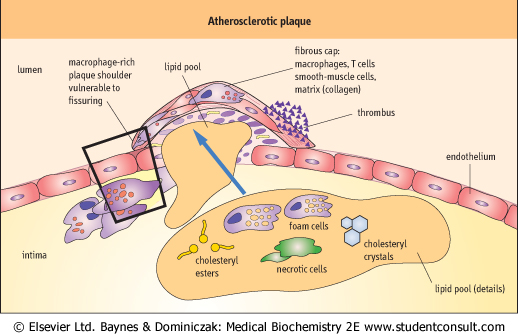
| As we already learned, the foam cells and the lipid pool form the center of developing atherosclerotic plaques. VSMC which had migrated into the intima synthesize new collagenous matrix which now forms a fibrous 'cap' over the lipid pool. This cap, however, is far from an inert structure: it contains VSMC themselves, macrophages, and T lymphocytes. The lipid pool and the cap constitute the mature atherosclerotic plaque, that on the one hand penetrates the arterial wall, and on the other obstructs the arterial lumen (Fig. 17.10). Parts of the more advanced lesions may also become calcified.
|
| A plaque decreases the blood flow to the area supplied by the affected vessel. In the coronary arteries, which supply the blood to the heart muscle, the obstruction by a plaque eventually results in the patient experiencing angina pectoris. If the narrowing occurs in the arteries supplying the legs, it causes intermittent claudication; if it is present in the carotid arteries it may cause the so-called transient ischemic attacks. A plaque may grow slowly and remain stable for long periods of time (in such case the severity of symptoms does not increase: a patient continues to have, for instance, a so-called stable angina). Extensive arteriosclerotic changes can also develop within the arterial wall without much affecting the lumen.
|
| page 239 | | | page 240 |
| Figure 17.10 Atherosclerotic plaque. The lipid center and fibrous cap are the main parts of a mature atherosclerotic plaque which emerges from the structurally changed vascular wall. The so-called vulnerable plaque ruptures easily. This figure illustrates areas vulnerable to breakage and shows the obstructing thrombus formed at the rupture site. |
| The plaque growth may be periodically accelerated by a cycle of plaque rupture and thrombosis. This is how it happens:
|
| Active macrophages and T lymphocytes preferentially reside at the edges of the plaque (Fig. 17.10). There, macrophages secrete enzymes which degrade extracellular matrix in the plaque cap: these enzymes belong to the metalloproteinase (MMP) family, which includes collagenases, gelatinases, and stromyelysin. In addition, T cells activated by macrophages secrete interferon-γ (IFN-γ) and pro-inflammatory cytokines IL-1, IL-2, and TNF-α. IFN-γ is particularly dangerous because it induces macrophage MMP expression. It also inhibits VSMC proliferation and their collagen synthesis, further weakening the cap. VSMC present in the most vulnerable edge regions of the plaque undergo apoptosis. All this creates a plaque that is prone to rupture.
|
| When such unstable plaque ruptures, it exposes its interior to the blood. The interior is highly thrombogenic due to the presence of the tissue factor, a small-molecular-weight glycoprotein that initiates the extrinsic clotting cascade (see Chapter 6). Tissue factor forms a complex with factor VII/VIIa and this in turn activates factors IX and X. Platelets are also activated, and the thrombus forms quickly on the surface of a ruptured plaque. A thrombus which completely occludes the arterial lumen causes tissue necrosis in the area supplied by the involved artery. In the heart this is a myocardial infarction, and in the brain, a stroke.
|
| Not all instances of plaque rupture result in dramatic, complete lumen blockages. However, even small haemorrhages and thrombi forming either within the plaque, or on its surface, accelerate expansion of the plaque. The plaque grows faster than VSMC proliferation-matrix synthesis mechanism alone would allow, and may worsen angina. Such stepwise growth is the main mechanism of plaque expansion.
|
|