The urea cycle and its relationship to central metabolism cycle and its relationship to central metabolism
|
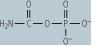
| The urea cycle (Fig. 18.7 and Table 18.2) was the first metabolic cycle to be well defined; its description preceded that of the TCA cycle. The start of the urea cycle may be considered the synthesis of carbamoyl phosphate from an ammonium ion and bicarbonate in liver mitochondria (Fig. 18.8). This reaction requires two molecules of ATP and is catalyzed by the enzyme, carbamoyl phosphate synthetase I (CPS I), which is found in high concentrations in the mitochondrial matrix.
|
| page 249 |  | | page 250 |
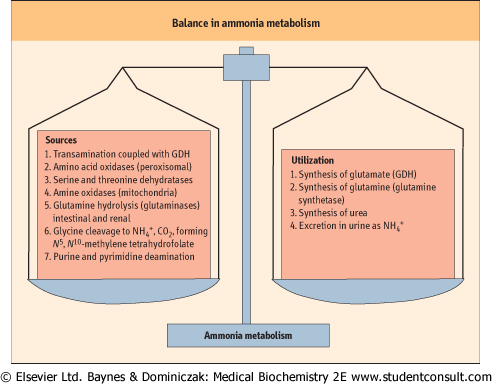
| Figure 18.6 Balance in ammonia metabolism. The balance between production and utilization of free ammonia is critical for maintenance of health. This figure summarizes the sources and pathways that use ammonia. Although most of these reactions occur in many tissues, urea synthesis is restricted to the liver. Glutamine and alanine function as the primary transporters of nitrogen from peripheral tissues to the liver. |
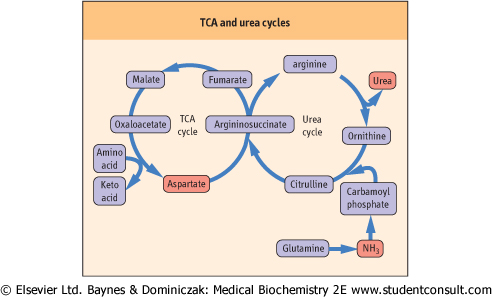
| Figure 18.7 TCA and urea cycles. Analysis of the urea cycle reveals that it is really two cycles: with the carbon flow split between the primary urea synthetic process and the recycling of fumarate to aspartate; the latter cycle occurs in the mitochondrion and involves parts of the citric acid cycle. |
| The mitochondrial enzyme, CPS I, is unusual in that it requires N-acetylglutamate as a cofactor. It is one of two carbamoyl phosphate synthetase enzymes that have key roles in metabolism. The second, CPS II, is found in the cytosol, does
not require N-acetylglutamate, and is involved in pyrimidine biosynthesis (Chapter 29).
|
| Figure 18.8 Synthesis of carbamoyl phosphate. Ammonia enters the urea cycle as carbamoyl phosphate, synthesized by carbamoyl phosphate synthetase in liver. |
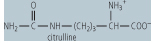
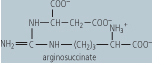
| Ornithine transcarbamoylase catalyzes the condensation of carbamoyl phosphate with the amino acid, ornithine, to form citrulline. In turn, the citrulline is condensed with aspartate to form argininosuccinate. This step is catalyzed by argininosuccinate synthetase and requires ATP; the reaction
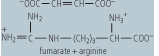
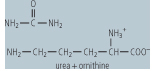
cleaves the ATP to adenosine monophosphate (AMP) and inorganic pyrophosphate (PPi) (2 ATP equivalents). The formation of argininosuccinate brings to the complex the second nitrogen atom destined for urea. Argininosuccinate is in turn cleaved by argininosuccinase, to arginine and fumarate. The arginine produced in this series of reactions is then cleaved by arginase, to a molecule of urea and one of ornithine. The ornithine can then be used to reinitiate this cyclic pathway, while the urea diffuses into the blood, is transported to the kidney, and excreted in urine. The net process of urea synthesis is summarized in Table 18.3.
|
| The urea cycle is split between the mitochondrial matrix and the cytosol
|
| page 250 | | | page 251 |
|
Table 18-2.
Enzymes of the urea cycle. |
| Body_ID: None |
| Enzymes of the urea cycle |
| Body_ID: T018002.50 |
| Enzyme | Reaction catalyzed | Remarks | Reaction Product |
| Body_ID: T018002.100 |
| Carbamoyl phosphate synthetase | formation of carbamoyl phosphate from ammonia and CO2 | fixes ammonia released from amino acids, especially glutamine, uses 2 ATP, located in the mitochondrion, deficiency leads to high blood concentrations of ammonia and related toxicity |
 |
| Body_ID: T018002.150 |
| Ornithine transcarbamoylase | formation of citrulline from ornithine and carbamoyl phosphate | releases Pi, an example of a transferase, located in themitochondrion, deficiency leads to high blood concentrations of ammonia and orotic acid, as carbamoyl phosphate is shunted to pyrimidine biosynthesis |
 |
| Body_ID: T018002.200 |
| Argininosuccinate synthetase | formation of argininosuccinate from citrulline and aspartate | requires ATP, which is cleaved to AMP + PPi - an example of a ligase, located in the cytosol, deficiency leads to high blood concentrations of ammonia and citrulline |
 |
| Body_ID: T018002.250 |
| Argininosuccinase | cleavage of argininosuccinate to arginine and fumarate | an example of a lyase, located in cytosol, deficiency leads to high blood concentrations of ammonia and citrulline |
 |
| Body_ID: T018002.300 |
| Arginase | cleavage of arginine to ornithine and urea | an example of a hydrolase, located in the cytosol and primarily in the liver, deficiency leads to moderately increased blood ammonia and high blood concentrations of arginine |
 |
| Body_ID: T018002.350 |
|
| Body_ID: T018002.400 |
Five enzymes catalyze the urea cycle in liver. The first enzyme, CPS-1, which fixes NH4+ as carbamoyl phosphate, is the regulatory enzyme and is sensitive to the allosteric effector, N-acetylglutamate.
|
|
Table 18-3.
Urea synthesis. |
| Body_ID: None |
| Urea synthesis |
| Body_ID: T018003.50 |
| Component reactions in urea synthesis |
| Body_ID: T018003.100 |
| CO2 + NH3 + 2 ATP | → | carbamoyl phosphate + 2 ADP + Pi |
| Body_ID: T018003.150 |
| Carbamoyl phosphate + ornithine | → | citrulline + Pi |
| Body_ID: T018003.200 |
| Citrulline + aspartate + ATP | → | arginosuccinate + AMP + PPi |
| Body_ID: T018003.250 |
| Arginosuccinate | → | arginine + fumarate |
| Body_ID: T018003.300 |
| Arginine | → | urea + ornithine |
| Body_ID: T018003.350 |
| CO2 + NH3 + 3 ATP + aspartate | → | urea + 2 ADP + AMP + 2 Pi + PPi + fumarate |
| Body_ID: T018003.400 |
| The mechanisms involved in ammonia toxicity - the encephalopathy in particular - are not well-defined. It is clear, however, that when its concentration builds up in the blood and other biological fluids, ammonia diffuses into cells and across the blood/brain barrier. This increase in ammonia causes an increased synthesis of glutamate from α-ketoglutarate and increased synthesis of glutamine. Although this is a normal detoxifying reaction in cells, when concentrations of ammonia are significantly increased, supplies of α-ketoglutarate in cells of the CNS may be depleted, resulting in inhibition of the TCA cycle and production of ATP. There may be additional mechanisms accounting for the bizarre behavior observed in individuals with high blood concentrations of ammonia. Either glutamate, a major excitatory neurotransmitter, or its derivative, γ-amino butyric acid (GABA), an inhibitory neurotransmitter, may also contribute to the CNS effects. |
| page 251 | | | page 252 |
| CARBAMOYL PHOSPHATE SYNTHESIS |
| The enzyme carbamoyl phosphate synthetase I (CPS I) is found in the mitochondrion and primarily in the liver; a second enzyme, CPS II, is found in the cytosol and in virtually all tissues. Although the product of both these enzymes is the same, namely carbamoyl phosphate, the enzymes are derived from different genes and function in urea synthesis (CPS I) or pyrimidine biosynthesis (CPS II), respectively. Additional differences between the two enzymes include their source of nitrogen (NH3 in the case of the former and glutamine in the latter) and their requirement for N-acetylglutamate (required by the former, but not by the latter). Under normal circumstances, CPS I and II function independently and in different cellular compartments; however, when the urea cycle is blocked as a result of a deficiency in ornithine transcarbamoylase, the accumulated mitochondrial carbamoyl phosphate spills over into the cytosolic compartment and may stimulate excess pyrimidine synthesis, which is reflected in a build-up of orotic acid in blood and urine. |
| An otherwise healthy, 60-year-old man noticed an occasional tremor in his left arm when relaxing and watching television. He also noticed occasional muscle cramping in his left leg, and his spouse noticed that he would occasionally develop a trance-like stare. A complete physical examination and consultation with a neurologist confirmed a diagnosis of Parkinson's disease. He was prescribed a medication that contained l-dihydroxyphenylalanine (l-DOPA) and a monoamine oxidase inhibitor (MAOI). l-DOPA is a precursor of the neurotransmitter dopamine, while monoamine oxidase is the enzyme responsible for the oxidative deamination and degradation of dopamine. His symptoms improved immediately, but he gradually experienced significant side effects from the medication, especially the occurrence of involuntary movements. |
| Comment. Parkinson's disease is caused by the death of dopamine-producing cells in the substantia nigra and the locus ceruleus. Although medication can markedly reduce the symptoms, the disease is progressive and may result in severe disability. Dopaminergic agonists often have side effects and also have limited effect on tremor so that other treatments such as deep brain stimulation or ablation are used in selected cases. Monoamine oxidase is also involved in deamination of other amine in the brain, so that MAOIs have many undesirable side effects. Transplantation of dopaminergic fetal tissue is a controversial experimental treatment at present (see Chapter 40). |
| HEREDITARY HYPERAMMONEMIA |
| An apparently healthy 5-month-old female infant was brought to a pediatrician's office by her mother, with a complaint of periodic bouts of vomiting and a failure to gain weight. The mother also reported that the child would oscillate between periods of irritability and lethargy. Subsequent examination and laboratory results revealed an abnormal electroencephalogram, a markedly increased concentration of plasma ammonia (323 mmol/L [550 mg/dL]; the normal range is 15-88 mmol/L [25-150 mg/dL]), and greater than normal concentrations of glutamine, but low concentrations of citrulline. Orotate, a pyrimidine nucleotide precursor, was found in her urine. |
| Comment. The infant was admitted to hospital and treated with intravenous phenylacetate and benzoate along with arginine. The infant improved rapidly, and was discharged from hospital on a low-protein diet with arginine supplementation. Subsequent biopsy of the patient's liver indicated that her hepatic ornithine transcarbamoylase activity was about 10% of normal. |
|
| The first two steps in the urea cycle occur in the mitochondrion. The citrulline then diffuses into the cytosol, where the
cycle is completed with the release of urea and the regeneration of ornithine. Ornithine must be transported back across the mitochondrial membrane to continue the cycle. It is also a precursor of putrescine, spermine and other polyamines found in the cell. These compounds bind to DNA and RNA, and function in cell growth and differentiation. Carbons from fumarate, released in the argininosuccinase step, may also re-enter the mitochondrion after hydration to malate and be
recycled via enzymes in the TCA cycle, to oxaloacetate and ultimately to aspartate (Fig. 18.7), thus completing the second part of the urea cycle. Because the same carbon skeleton of ornithine that begins the cycle is returned after the arginase reaction, this pathway is sometimes also referred to as the ornithine cycle. Urea synthesis occurs virtually exclusively in the liver and the role of the enzyme, arginase, in other tissues is probably related more closely to ornithine requirements in those tissues than to the production of urea.
|
| Regulation of the urea cycle
|
| The primary regulation of the urea cycle appears to be through the control of the concentration of N-acetylglutamate, an essential allosteric activator of CPS-I. High concentrations of arginine stimulate N-acetylation of glutamate. Concentrations of urea cycle enzymes also increase or decrease in response to a high- or low-protein diet. Urea synthesis and excretion is also decreased, and NH4+ excretion increased, during acidosis.
|
| page 252 | | | page 253 |
| SCREENING FOR DEFECTS OF AMINO ACID METABOLISM IN NEWBORNS |
| In most developed countries today, a spot of the blood of newborn infants is routinely collected on filter paper and tested for a series of compounds which are markers of inherited metabolic disease. The number of markers analyzed may vary from state to state in the US, but generally ranges from 10 to 20. Because of the need for rapid screening, small sample size, and reduced cost, older methodology is rapidly being replaced by technology which uses electrospray tandem mass spectrometry to measure the level of multiple markers simultaneously. The speed and high throughput capacity of this technology allows rapid screening of 20 or more markers from dried blood spots and the identification of infants who are potential victims of these inborn errors of metabolism. This technology can also be applied to urine samples. |
|
| Defects in any of the enzymes of the urea cycle (Table 18.2) have serious consequences. Infants born with defects in any of the first four enzymes in this pathway may appear normal at birth, but rapidly become lethargic, lose body temperature, and may have difficulty breathing. Blood concentrations of ammonia increase quickly, followed by cerebral edema. The symptoms are most severe when early steps in the cycle are
affected. However, a defect in any of the enzymes in this pathway is a serious issue and may cause hyperammonemia and lead rapidly to CNS edema, coma and death. Ornithine transcarbamoylase is the most common of these urea cycle defects and shows an X-linked inheritance pattern. The remainder of the known defects associated with the urea cycle are autosomal recessive. A deficiency of arginase, the last enzyme in the cycle, produces less severe symptoms, but is nevertheless characterized by increased concentrations of blood arginine and at least a moderate increase in blood ammonia. In individuals with high blood concentrations of ammonia, hemodialysis must be used, often followed by intravenous administration of sodium benzoate and phenyllactate. These compounds can condense with glycine and glutamine, respectively, trapping ammonia in a nontoxic form that can be excreted in the urine. The enzymes of the urea cycle are listed in Table 18.2.
|
|