| DNA cloning by the polymerase chain reaction (PCR)
|
| The use of PCR for the amplification of DNA in vitro has revolutionized molecular biology; it is a method of copying a single template DNA molecule
|
| PCR is a simple and quick means of generating up to 109 copies of a single template DNA molecule within hours. A standard PCR reaction requires the following:
|
- DNA template: DNA containing the sequence to be amplified;
- amplimers: small oligonucleotide primers that will hybridize with complementary DNA sequences both upstream and downstream of the desired sequence region and act as a starting point for the synthesis of the newly amplified DNA strands;
- polymerase enzyme: typically a heat-stable polymerase that will catalyze the formation of the DNA during the amplification reaction;
- dNTPs: these are of course essential for the synthesis of the new DNA strands by the polymerase.
|
| page 481 |  | | page 482 |
|
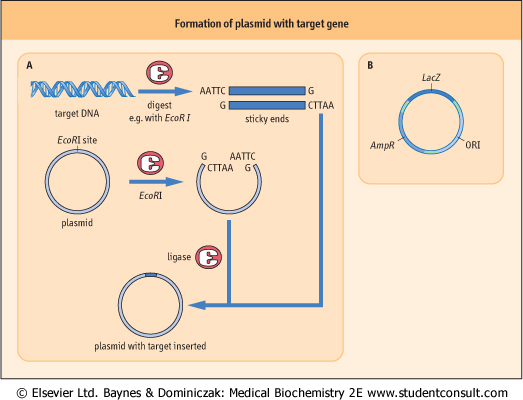
Figure 34.9 Formation of a plasmid containing a target gene for cloning. (A) DNA containing the target gene is digested with a restriction enzyme that will produce 'sticky ends', e.g. EcoRI. The plasmid also has a restriction site for EcoRI, so that when digested with EcoRI it becomes a linear DNA strand with 'sticky ends' complementary to the target. Upon ligation the target and vector form a recombinant molecule. (B) Structure of a typical plasmid. The plasmid contains a gene conferring resistance to the antibiotic ampicillin (AmpR), and a portion of the LacZ gene that encodes the N-terminal fragment of the enzyme β-galactosidase. The E. coli strain used for this work has a defective β-galactosidase enzyme which is complemented by the plasmid LacZ gene product to form a functional enzyme. Within the plasma LacZ gene segment is a polylinker region containing approximately 10 restriction-enzyme recognition sites, which serve as sites for insertion of target DNA. The plasmid also contains ORI, the site for origin of DNA replication. (AmpR), and a portion of the LacZ gene that encodes the N-terminal fragment of the enzyme β-galactosidase. The E. coli strain used for this work has a defective β-galactosidase enzyme which is complemented by the plasmid LacZ gene product to form a functional enzyme. Within the plasma LacZ gene segment is a polylinker region containing approximately 10 restriction-enzyme recognition sites, which serve as sites for insertion of target DNA. The plasmid also contains ORI, the site for origin of DNA replication. |
| VECTOR SYSTEMS FOR CLONING LARGE DNA FRAGMENTS |
| One critical factor in recombinant DNA manipulations is the size of the target DNA. Conventional bacterial plasmids, although convenient to work with, are limited in the size of insert they can accept; 1-2 kb is the common size of the insert, with an upper limit of 5-10 kb. Some modified plasmid vectors called cosmids can accept larger fragments up to 20 kb. Another commonly used vector that has the ability to accept larger DNA fragments is the bacteriophage lambda (λ). This viral particle contains a double-stranded DNA genome packaged within a protein coat. The λ-phage can infect E. coli cells with high efficiency and introduce its DNA into the bacterium. Infection leads to the replication of viral DNA and the synthesis of new viral particles, which can then lyse the host cell and infect neighboring cells to repeat the process. The viral DNA is then re-isolated to obtain the recombinant DNA. |
| Larger inserts can be cloned by using modified chromosomes from either bacteria (bacterial artificial chromosomes, BACs), or yeast (yeast artificial chromosomes, YACs). Such vectors can accommodate DNA fragments up to 1-2 Mb. BACs have been particularly important in putting together the sequence of the human genome. |
| page 482 | | | page 483 |
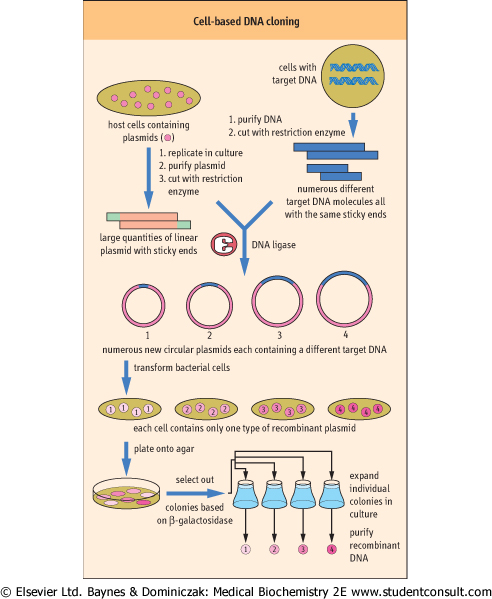
| Figure 34.10 Cell-based DNA cloning. An example of cloning genomic DNA using bacterial cells. In general each transformed bacterium will take up only a single plasmid molecule. Therefore individual bacterial colonies will contain many identical copies of just one particular recombinant DNA. |
| PCR consists of a series of programmed temperature changes that serve to bring about a round of DNA synthesis
from the primers (Fig. 34.11). The average PCR involves about 30-35 cycles of reactions, which provide sufficient DNA, as much as a billion copies of the original template, so that it can be visualized following electrophoresis on an agarose gel.
|
| CELL-BASED CLONING IS USED TO PRODUCE CLINICALLY IMPORTANT PROTEINS |
| A 13-year-old girl was admitted with dehydration, vomiting and weight loss. Her blood glucose level was 19.1 mmol/L (344 mg/dL) and she had ketonuria. A diagnosis of type 1 diabetes mellitus was made. She was started on recombinant human insulin, was rehydrated, and made a prompt recovery. |
| Comment. Prior to the advent of recombinant DNA technology, insulin therapy involved the use of animal insulins, most commonly pork or beef, which were chemically similar, but not identical, to human insulin. As a result of these differences, animal insulins often led to the development of antibodies, which reduced the efficacy of the insulin and could lead to treatment failures. |
| Insulin was the first biologically important human molecule to be produced by means of recombinant DNA technology. Following the cloning of the human insulin gene, large-scale production of pure human insulin was possible by inserting the cloned gene into a cell-based amplification system. Large amounts of insulin gene copies were produced, which were then expressed in either bacteria or yeast, and the resulting purified insulin was made available for use in treatment of diabetic patients. |
| One crucial step in the process is the use of cDNA as a template for transcription and translation of the desired gene. By using insulin mRNA as a template, human insulin cDNA can be generated by reverse transcription (formation of a complementary DNA strand using mRNA as the template) whose product is a DNA molecule that contains all the coding region of the insulin gene without any introns. By this means, human recombinant insulin has almost totally replaced animal insulin in the treatment of diabetes. Other important recombinant human peptides used clinically include growth hormone, erythropoietin and parathyroid hormone. |
The cycle of the PCR comprises three steps that are repeated continuously by varying temperature in a cyclic fashion:
- denaturation: heating of the reaction to approximately 95°C to ensure template and primers are single stranded;
- annealing: cooling of the mixture to allow heteroduplexes of primer and template to form. The temperature for this is based on the Tm of the expected duplex (typically in the 50 to 65°C range);
- elongation: DNA polymerase elongates the newly synthesized DNA strand from the site of the annealed primer along the template strand (typically about 70°C).
|
| page 483 | | | page 484 |
| The practical development of PCR was made possible by the discovery of heat-stable DNA polymerases that can withstand the temperature of the denaturation step and still
retain enzymatic activity, e.g. Taq and Pfu polymerases. These enzymes were isolated from organisms that live and reproduce in hot springs and geysers.
|
| The ability of PCR to amplify selectively a single template DNA relies on some prior knowledge of the nucleotide sequences in the regions flanking the target sequence
|
| If the sequences in the regions flanking the target sequence are known, oligonucleotide primers are synthesized that are complementary to:
|
- the region on the sense (top) strand upstream of the target region; and
- the region on the antisense (lower) strand downstream of the target region (Fig. 34.11).
|
| It is essential that the sequence of the primers be unique to the target DNA being amplified. If, for instance, the downstream primer contains sequences that contain tandem repeats of bases, e.g. TGTGTG, the specificity of the amplification may be reduced as such repetitive sequences occur widely throughout the genome, and the amplified sequence may contain many unwanted products.
|
|
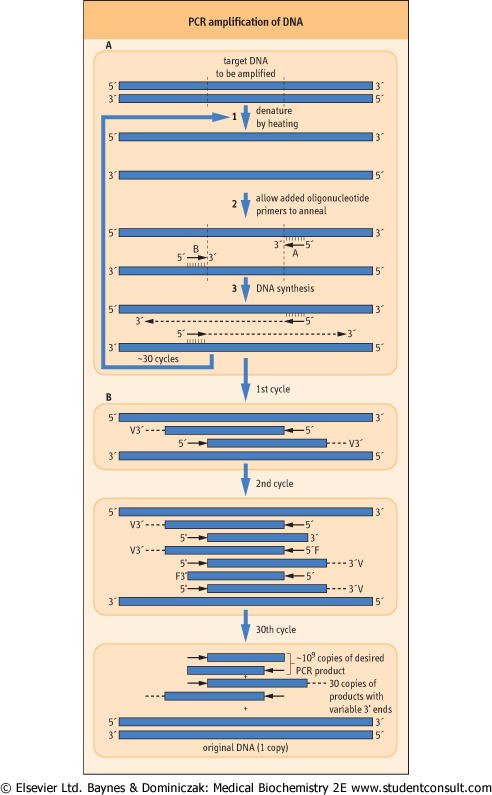
| Figure 34.11 Polymerase chain reaction (PCR) amplification of DNA. (A) Cycle 1. The target DNA is heated to 94°C to ensure complete denaturation. The mixture is then cooled to allow the oligonucleotide primers to hybridize to the complementary sequence in the target DNA. Specificity is provided by using two primers that anneal to sequences on opposite strands at opposite ends of the sequence to be amplified. In the presence of dNTPs, Taq polymerase catalyzes the elongation, beginning at the 3'end of the primers. Two new duplex DNA molecules: one generated by each primer on its respective template. (B) Subsequent cycles. In the second cycle, products from the first cycle become templates. These templates have primers at their 5'-ends. Accordingly, the DNA made on them stops at the end of the primers used in the first cycle. From now on, molecules will accumulate during each cycle that have both ends defined by the pair of primers used. These accumulate exponentially and soon far outnumber the initial input DNA template. After 30 cycles, the original template and a trivial number of the variable-ended strands copied directly from it remain, but the number of amplified target molecules enclosed by the primers approaches 109. |
| page 484 | | | page 485 |
| Advantages and disadvantages of PCR cloning
|
| PCR is a quick, simple and robust technique, but prior sequence knowledge is needed, only a relatively small target DNA can be amplified, and DNA replication may be inaccurate
|
PCR has three principal advantages over cell-based cloning:
- time: by using PCR, a single template strand can be amplified to produce a detectable product very rapidly. Each step takes a few minutes or less, and 30 cycles of this PCR process can be completed in at most a few hours. Cell-based cloning is more expensive and may require days or weeks to produce a similar yield of DNA;
- sensitivity: PCR can amplify a DNA template to usable amounts if only a single copy of the template DNA is present. Indeed, PCR protocols have been developed to allow amplification of target DNA from a single cell. This is why PCR has found such a prominent place in forensic applications and even in paleontology;
- robustness: PCR is able to amplify DNA that is often badly degraded by time or the elements, or is present in previously inaccessible sites, e.g. formalin-fixed tissue. Only a few copies of an unmodified, relatively short sequence to be amplified need to remain intact.
|
Despite the apparent attraction of PCR, it is not without fault and, depending on the application of the method, it may be an unacceptable means of amplifying DNA. The drawbacks of PCR are as follows:
- prior sequence knowledge: to perform PCR, flanking primers must be synthesized. For this to happen, the sequence of the target DNA, or at least its flanking regions, must be known. This may require prior cell-based cloning to derive the flanking nucleotide information. With the virtual completion of the human genome sequence, this issue is less important than in prior years;
- small target DNA amplification: PCR will reliably amplify target sequences up to about 5 kb, although products of 200-1000 bases are most readily amplified. However, larger products have been amplified recently by so-called 'long PCR' methods, allowing sequences up to 20 kb or more to be amplified;
- DNA replication may be inaccurate: errors can be introduced during DNA polymerization by Taq polymerase since this enzyme lacks proof-reading activity. In a standard PCR reaction, up to 40% of the synthesized products will have some error in the nucleotide sequence introduced by the polymerase enzyme, usually only a single nucleotide substitution. Therefore, the final reaction mixture will contain numerous copies of the target DNA, which are almost but not quite the same, and the substituted nucleotide may be different in different amplified molecules. Similarly, if there is any contamination of the DNA source by other DNAs that may contain the target sequence (such as with forensic material), unwanted amplification may occur.
|
| Thus, it is safe to say that PCR provides a simple, fast and very efficient means of amplifying DNA that is partly or fully characterized, is of relatively small size and may be present in small amounts or in a poor condition. However, it is a less useful technique if the objective is to amplify DNA accurately without introducing sequence errors or if the target DNA sequence is large.
|
|