| Bcl-2 is a key antiapoptotic protein
|
| page 592 |  | | page 593 |
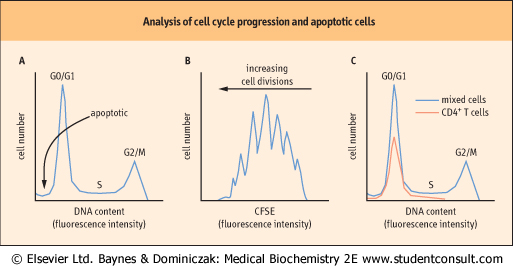
| Figure 41.8 Analysis of cell cycle progression and apoptotic cells. (A) Measurement of DNA content in PI- or DAPI-stained cells by flow cytometry allows assessment of the cell cycle status of individual cells in a population. Tetraploid = G2/M, diploid = G0/G1, intermediate = S phase; subdiploid DNA content indicates cells undergoing apoptosis. (B) When CFSE-stained cells divide, CFSE is shared equally between daughter cells, reducing the fluorescence intensity of daughter cells by half with each division. Flow cytometry reveals discrete cell subpopulations representing successive generations of cells. (C) Surface staining of cells with a fluorescent antibody specific for the T helper cell marker CD4 suggests that CD4+ T cells in this mixed population of cells are arrested in G0. |
| GENETIC MUTATIONS UNDERLYING DEVELOPMENT OF A COLORECTAL CANCER |
| Screening of colorectal cancer cells with DNA probes specific for known proto-oncogenes and tumor suppressor genes showed that 50% of these tumors had an activating point mutation in a Ras oncogene, and 75% of these cancers had an inactivating mutation in p53. Moreover, studies of familial adenomatous polyposis (FAP; see below) have shown that more than 70% of colon cancers have a mutation in the adenomatous polyposis coli (APC) gene. Indeed, it now seems clear that mutations inactivating APC may represent an early stage in the development of a colon cancer cell, because APC mutations can be detected at the same frequency in small benign polyps as in large malignant tumors. These APC mutations appear to promote the transformation of normal epithelium through a stage of hyperproliferative epithelium to early adenoma. The next stage seems to involve the activating mutation of Ras (rarely found in small polyps, but common in large polyps), which induces changes in cell differentiation typical of cell transformation (intermediate adenoma) and which allow the cells to grow in an anchorage-independent manner in culture. These changes are followed by a mutation in a tumor suppressor gene called 'deleted in colon carcinoma' (DCC), to generate a late adenoma that is converted to an adenocarcinoma by the subsequent loss of p53. Loss of the DNA-damage sensing function of p53 allows the cells to accumulate, at a rapid rate, the further mutations required for metastasis. |
| Comment. Although these mutations appear to be the rate-limiting steps in a large number of colorectal cancers, it should be noted that other sequences of mutations can also generate this disease. Indeed, in about 15% of cases, patients with a hereditary syndrome called hereditary nonpolyposis colorectal cancer (HNPCC) have a mutation of the HNPCC gene that normally acts to repair mismatches in the nucleotide sequences of the DNA double helix. Thus, in such patients, the likelihood of the development of colorectal cancer is enhanced, as the HNPCC mutation decreases the fidelity of DNA replication and hence increases the tendency of the cells to undergo mutation. |
| The Bcl-2 gene family comprises structurally related pro-teins that form homo- or heterodimers and act either to
promote or to antagonize apoptosis by modulating caspase activation.
|
| Bcl-2 was initially discovered as a chromosomal translocation in a follicular B cell lymphoma, which was found to contribute to neoplastic growth by inhibiting normal B cell apoptosis. The molecular mechanisms underlying Bcl-2-mediated survival signals are as yet poorly defined, but it appears that Bcl-2, which is located on the outer mitochondrial membrane, may act by preventing leakage of cytochrome C from the mitochondria and, hence, activation of effector caspases (see Fig. 41.7).
|
|