These experiments are designed to be carried out in 36 hours (6 x 6).
In the first 2-3 weeks, several simple coordination complexes are prepared. During this
period as well, some qualitative tests are done on first row transition metal ions.
In the fourth week, a kinetics experiment on the acid hydrolysis of a metal complex is
carried out as a group exercise. Each student is assigned a variation such as initial
concentration of complex, temperature, ionic strength or [H+] concentration.
At the end of the session, all rate constants are pooled so that extensive analysis can be
done.
In the last 2 weeks a number of spectroscopic investigations are performed, including: IR,
UV/Vis and a determination of a magnetic moment. This is done using a rotation scheme to
accommodate the time required at each instrument and to give each student the opportunity
to record their own spectra.
The preparation of ferrocene is carried out during this 2 week period as well.
It is intended that the laboratory experiments will reinforce the lecture material and give students practice at assigning spectral bands and interpreting magnetic properties as well as giving an introduction to the study of reaction kinetics.
Experiment 1 :
Preparation of some 2,4-pentanedione (acac) complexes, either:
i) VO(acac)2, VO(acac)2.pyr and Co(acac)3 ii) Cr(acac)3 and trans Ni(acac)2·2H2O or iii) Mn(acac)3 and Fe(acac)3
The final reports should include the Vis and IR
spectroscopic analysis and Gouy or Evans Method determination
of the magnetic moment. It is expected that the discussions will be based on the
appropriate Orgel diagram and include an assessment of how the measured values compared
with what was expected.
Experiment 2:
Preparation and analysis of potassium trisoxalatoiron(III) trihydrate
Experiment 3:
Preparation and aquation of trans-dichlorobis(1,2-diaminoethane)cobalt(III) chloride
Experiment 4:
Preparation of some Werner complexes.
Experiment 5:
Preparation and reactions of ferrocene.
Experiment 6:
Qualitative Tests
All samples should be submitted in properly labelled (name, sample, weight etc.)
containers for marking, together with your reports.
You are reminded that the Practical work contribution is worth 20% of the total for this
course and that students not performing satisfactorily may be required to sit a Practical
Examination. A pass in the practical is essential for passing the course overall.
To 1 g of V2O5 in a 200 cm3 beaker, add 6 cm3 of 4 mol
dm-3 sodium hydroxide and 25 cm3 of distilled water. Heat the
mixture almost to boiling to dissolve as much of the solid as possible. Cool the solution
to room temperature and then add 1 g of sodium metabisufite, Na2S2O5
rapidly followed by 12 cm3 of 2 mol dm-3 of H2SO4,
while stirring the solution. Then boil to remove excess SO2. Filter the hot
solution through a sintered glass crucible (#3) into 3 cm3 of pentane-2,4-dione
(acetylacetone). Carefully neutralise the mixture by dropwise addition of sodium carbonate
solution (4 g Na2CO3 dissolved in 25 cm3 of water).
Cool the neutral mixture and filter at the pump the precipitate which has formed. Dry the
crude product by suction in air. Save some for the next preparation and recrystallise the
rest.
1 g can be recrystallised by dissolving it in the minimum of boiling chloroform and
precipitating the pure product by adding an equal volume of petroleum ether (60/80). (Care!
inflammable - heat on a steam bath). Filter off the product at the pump and dry it by
suction in air. Record the yield.
An IR spectrum is available, however it shows
additional peaks from CO2 and water vapour which were not removed by background
subtraction. The FTIR still needs degassing and new dessicant it seems, after being
switched off for 18 months!!
Suspend 0.5 g VO(acac)2 in a mixture of 5 cm3 of ether and 1 cm3 of pyridine. Gently warm the suspension on a steam bath in a fume cupboard for 10 minutes. Filter off the olive-green product and wash with 3 x 3 cm3 of ether to remove the remaining traces of excess pyridine. Record the yield.
Dissolve 5.3 g of chromium(III) chloride hexahydrate in 80 cm3
of water in a 100 cm3 flask containing 5 g urea. Transfer the contents to two
120 cm3 Teflon bombs. Add 6 cm3 of
acetylacetone to each bomb and securely seal them and then place them in the microwave
oven. Set the timer for 5 minutes and watch the container throughout the reaction. If
after 5 minutes no precipitate is observed, continue heating for another 2 minutes. Remove
the bombs and cool them before opening. Transfer the contents back to your flask and then
collect the red precipitate by filtration (use a sinter # 3). Wash the crystals with small
portions of cold water and dry by sucking air through them. Weigh the dried product. |
Dissolve 0.6 g of manganese(II) chloride and 1.6 g of NaOAc·3H2O in 25 cm3 of water. Add 3 cm3 of acetylacetone slowly with stirring. Treat the resultant two-phase system with potassium permangante solution (1.2 g in 6 cm3 of water) and after a few minutes add, in small amounts with stirring, sodium acetate solution (1.6 g NaOAc·3H2O in 6 cm3 water). Heat the solution to about 60°? C for 10 minutes, cool in ice-cold water and filter at the pump. Wash the product with ice-cold water and small quantities of acetone to facilitate drying. Dry at the pump and determine the yield.
Dissolve 5 g of ferric sulfate in 25 cm3 of water and add 4 cm3
of 2,4 pentanedione. Dissolve 3.5 g of sodium acetate in 25 cm3 of water.
Slowly add the second solution to the first, stirring continuously. Filter off the red
crystals and air dry them. Weigh the dried product.
An IR spectrum is available.
A mixture of 2.5 g CoCO3 and 20 cm3 of acetylacetone is heated on a steam bath to approximately 85°? C. On adding 30 cm3 of 10% H2O2 dropwise to the mixture it becomes green in colour. When effervescence ceases, the reaction is complete. The flask is chilled in ice and the product filtered (use a #3 sinter) The product is then washed with cold ethanol and dried at 110 C for 15 min. Weigh the dried product.
Dissolve 2 g NiOAc·4H2O in the minimum quantity of hot ethanol. Add to this slowly, with stirring, 5 cm3 of acetylacetone in 20 cm3 of ethanol. Cool in an ice-bath and collect the green crystals. Weigh the dried, crude product and recrystallise it from ethanol.
An IR spectrum is available, however it shows additional peaks from CO2 and water vapour which were not removed by background subtraction. The FTIR needs further degassing and some new dessicant after being switched off for 18 months!!
Mark the level of 45 cm3 water in a 250 cm3 beaker. To a well-stirred solution of 5 g of ferrous ammonium sulfate in 20 cm3 of warm water containing 1 cm3 of dilute sulfuric acid in the beaker, add a solution of 2.5 g of oxalic acid dihydrate in 25 cm3 of water. Slowly heat the mixture to boiling (beware of bumping) then allow thc yellow precipitate to settle. Decant the supernatant through a Buchner funnel making sure it has a properly fitted filter paper. Add 15 cm3 of hot water to the solid, stir and filter. Drain well and then transfer all the precipitate from the paper back into the beaker with 10 cm3 hot water. |
Add 3.5 g solid potassium oxalate monohydrate and heat to approximately 40°? C.
Add slowly, using a dropper, 9 cm3 of "20 vol" hydrogen peroxide. (If
the precipitate looks yellowish, not brown and settles readily, decant the supernatant,
add a solution of 0.2 - 0.4 g potassium oxalate monohydrate in 1 - 2 cm3 water
and then hydrogen peroxide dropwise until the precipitate dissolves. Then add the
previously decanted supernatant). Heat to boiling, and add a solution of 2 g of oxalic
acid dihydrate in 30 cm3 of water in portions, add 20 cm3 initially,
then if the brown precipitate still remains, add more solution little by little until it
all dissolves. Boil the clear solution down to a volume of 40 - to 50 cm3,
filter through a Buchner funnel with well fitting paper and add 95% ethanol slowly until a
precipitate starts to form (~30cm3). Redissolve any crystals by heating (beware
of fire) and leave to crystallise.
Filter and wash the crystals on the Buchner with a 1:1 ethanol / water mixture and finally
with acetone, (beware fire again). Dry in the air and weigh. The complex is
photosensitive and should not be exposed to light unnecessarily. Store in a sample bottle
wrapped in foil.
An IR spectrum is available.
The iron(III) complex is first decomposed in hot acid solution and the free oxalic acid
is titrated against standard (0.02 M) potassium permanganate solution. No indicator is
required.
In duplicate, weigh accurately about 0.2 g of the potassium trisoxalatoferrate(III)
complex prepared previously. Boil the sample with 50 cm3 of 1 M sulfuric acid
in a conical flask. Allow the solution to cool to about —60°? C and titrate
slowly with the otassium permanganate solution provided (which you will need to
standardise). Continue until the warm solution retains a slight pink colouration after
standing for about 30 sec.
Calculate the percentage by weight of oxalate in the complex, compare this with the
theoretical value and thus obtain the percentage purity of the complex.
MnO4- + 8H+ + 5e- -> Mn2+ + 4H2O C2O42- -> 2CO2 + 2e-
Prepare duplicate solutions containing 0.2 g accurately weighed of your sample in 15 cm3
of dilute sulfuric acid. Dilute the solutions to 50 cm3 with distilled water
and expose them to sunlight for one hour (note carefully what happens). Titrate with your
standardised permanganate to determine the amount of reducing agent present.
Expose a small portion of your product to sunlight for several hours. Make sure that the
crystals have been ground to a fine powder and that you periodically stir the crystals so
that all the sample gets exposed equally to the sunlight. Perform the following tests on
samples of both irradiated and unirradiated complex:
Dissolve your sample in dilute sulfuric acid and divide the solution into three.
1) treat with a freshly prepared solution of potassium ferrocyanide. 2) treat with a freshly prepared solution of potassium ferricyanide. 3) treat with a solution of potassium thiocyanate.
Record carefully all observations.
Coordination complexes of cobalt(III) undergo ligand exchange or substitutions slowly
as compared to many other transition metal compounds. Their slow reactions have made them
suitable for kinetic investigations of their reaction mechanisms. The present experiment
involves a kinetic study of the acid hydrolysis of trans-[Co(en)2Cl2]Cl,
whereby the probable mechanism of the octahedral cobalt(III) substitution can be
determined. The reaction will be conducted such that each student will investigate one of
the following variations: pH, temperature, concentration and ionic strength. The entire
class will collate these results which can be used to show the effect of these variations
on the aquation.
Complexes of this type undergo aquation in a step-wise fashion according to the equations:
trans-[Co(en)2Cl2]+ + H2O -> trans-[Co(en)2(H2O)Cl]2+ + Cl-
trans-[Co(en)2(H2O)Cl]2+ + H2O -> Co(en)2(H2O)23+ + Cl-
where the first step is the one to be measured quantitatively in this experiment.
In principle, two fundamentally different mechanisms are possible for these reactions; a
dissociative or associative mechanism. The kinetic rate laws expeected for these two types
of mechanism are:
dissociative: Rate =k1[complex]
associative: Rate =k2[complex][H2O]
=kobs[complex]
that is they are dependent only on the concentration of the complex and are first order. This observation, however, furnishes no information as to the role played by the water and does not give any information about the molecularity of these reactions. Nevertheless the way in which the rate constant is affected by various changes in the nature of the complex ion is expected to give us information about the mechanism. It has been found that increasing chelation such as replacing two NH3 ligands by one ethylenediamine slows down the rate of acid hydrolysis. Allowing for the chelation effect the divalent monochloro complexes react about 100 times slower than the univalent dichloro complexes. |
CoCl2·6H2O (2 g) is dissolved in 2 cm3
of water in a beaker and 1 cm3 of 1,2-diaminoethane (en) in 5 cm3 of
water is slowly added cautiously and with stirring. The solution is cooled in an ice bath
to 5° C and 2 cm3 of H2O2 (30%) is slowly added
while maintaining the temperature at 5° C. [ CAUTION: Keep H2O2
off the skin and eyes!]. Then the solution is gently warmed to about 60°-70°
C for 15-20 minutes.
Concentrated HCl (4 cm3) is then added, and the solution evaporated on a steam
bath with occasional stirring to about 10 cm3. After cooling the solution in an
ice-bath, 3 cm3 of ethanol is added and the mixture cooled for a further 10
minutes. The resultant green crystals of trans-[Co(en)2Cl2]Cl·HCl·2H2O
are filtered onto a sintered glass Buchner funnel, washed with ethanol and sucked dry.
An IR spectrum is available.
To drive off the HCl of crystallization, place the dark green crystals in a small beaker
containing 5 cm3 of methanol and vigorously stir these with a glass stirring
rod. Transfer the resultant slurry to a large test tube. Place the test tube in a beaker
of water, then heat the water (gently at first) until the methanol has evaporated and no
more HCl gas is driven off. (The presence of HCl can be tested by holding a piece of moist
litmus paper at the mouth of the test tube).
Boiling for 15 minutes after the methanol has evaporated is usually sufficient.
Trans-[Co(en)2Cl2]Cl thus obtained is a light green powder.
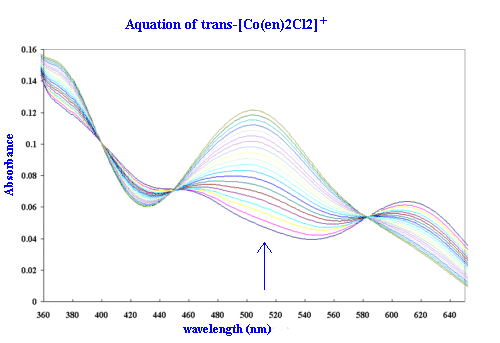
The visible spectrum of the complex ion, trans-[Co(en)2Cl2]Cl, has been investigated thoroughly. The peak at 625 nm is clearly defined with an extinction coefficient of 3.33m2mol-1. The trans configuration is well characterized by its shoulder at 440 nm. In acidic solution the trans-dichloro complex gradually converts to a mixture of 35% cis- and 65% trans-[Co(en)2(H2O)Cl]2+. By repetitively scanning the spectrum, this conversion is revealed by the presence of three poorly defined isosbestic points (588 nm, 448 nm and 408 nm).
Kinetic runs for the acid hydrolysis of the trans-[Co(en)2Cl2]Cl
complex are to be recorded at 515 nm where the increase in absorbance shows a maximum
change to occur between the reactant and product. The first
and last scan from a typical student run are
available for comparison.
Each student will be assigned to study the effect of one variation on the observed rate
constants, from the following parameters:
(a) Complex concentration (3mM to 15 mM). (b) Hydrogen ion concentration (0.1 M to 0.5 M). (c) Ionic strength (0.1 to 0.5 M). (d) Temperature (25° to 45° C).
A typical run should be done as follows.
Add required volumes of stock HNO3 and NaNO3 solutions to a 50 cm3 volumetric flask and add distilled water to make it about 80% full. The flask is then immersed in a thermostatic water bath for temperature equilibration. Weigh the required amount of the complex on an analytical balance and dissolve the complex with a small volume of distilled water in a beaker. Transfer the solution quantitatively to the thermostated 50 cm3 flask and make up to the mark with distilled water. Transfer immediately about 3 cm3 of the solution from the flask to a 1 cm glass or plastic cell, start your stop watch and place the cell in the cell holder of the spectrophotometer. Read and record the absorbance at 10 minute intervals for at least 2 hours. The absorbance reading at infinite time can be taken after 5 hours. Alternatively the reading at infinite time can be obtained by first warming a sample of the mixture on a water-bath for 5 minutes and then placing in the spectrometer.
For first order kinetics the following rate expression is expected:
ln( (Ainf - At) / (Ainf - A0) ) = kt
where k is the rate constant
A0 is the initial Absorbance
Ainf is the Absorbance at infinite time
and At is the Absorbance at any time, t.
Calculate the pseudo first order rate constants by plotting ln(Ainf - At)vs
time, t, in sec.
1. Draw the two structures of the cis and trans isomers of [Co(en)2Cl2]Cl.
Which, if any, of these geometric isomers is potentially resolvable into optically active
isomers?
2. The green product first isolated, is best represented as trans-[Co(en)2Cl2]+[H5O2]+2Cl-.
This contains an example of a hydrated proton, where the O-H-O moiety is linear and the
O-O separation is 200 pm. Draw the likely structure of the cation. Give examples of other
known hydrated proton structures.
For a clue see here.
3. In the complex [Co(NH3)5Cl]2+ increasing chelation
such as replacing the two NH3 ligands by one ethylenediamine slows down the
rate of acid hydrolysis. Why?
4. Why should the rate of hydrolysis of [Co(en)2Cl2]+ be
100 times faster than that of [Co(en)2Cl(H2O)]2+ ?
5 Tabulate all the experimental data collected during the class and comment on the effect
of (a) pH, (b) temperature and (c) ionic strength on the rate of hydrolysis.
6. Calculate DH# and DS# values from
your temperature dependence data and compare these values with the values obtained for
other chloroamine complexes of cobalt(III).
F. Basolo and R.G. Pearson, Mechanism of Inorganic Reactions, 2ed., John Wiley and
Sons, New York, 1967, p162.
B. Douglas, D.H. Daniels and J.J. Alexander, Concepts and Models of Inorganic Chemistry,
2nd Edition, John Wiley and Sons Inc., New York, 1983, pps 361-363.
The first step involves the preparation of cyclopentadiene. This is obtained by
cracking the dimer, dicyclopentadiene, by distillation (BP~42°C). This corresponds
to a retrograde Diels-Alder reaction and the cyclopentadiene monomer thus obtained
dimerises slowly (t½~12 hours at room temperature) and should be used without
delay. It is also highly flammable and should be stored in ice.
The next step makes use of the fact that alkali metal hydroxides are able to deprotonate
the cyclopentadiene when used in a non-hydroxylic solvent in which they are essentially
insoluble, in this case DMSO.
The final step is the reaction with the ferrous chloride which should be done quickly to
avoid too much air affecting the reaction.
It is essential to efficiently coordinate these steps of the procedure if good yields are
to be obtained. The experiment MUST be carried out in a fume hood.
The equipment for cracking the dicyclopentadiene will be set up by a demonstrator. Each
student requires 4 cm3 of the cyclcopentadiene. Take two boiling tubes each
fitted with a cork. Into the first place 6 cm3 of dimethyl sulfoxide and 3g of
KOH pellets which have been finely ground in a mortar and pestle. (Caution - If KOH is
spilt on the bench, it needs to be cleaned up immediately).
Into the second boiling tube, place 10 cm3 of dimethyl sulfoxide and 2 g of
anhydrous ferrous chloride.
The air in the boiling tubes must now be replaced by dinitrogen and the tubes shaken.
Consult the demonstrator for the use of the dinitrogen gas cylinder.
The next two operations must be carried out quickly so as to prevent too much air getting
into the boiling tubes. Add the 4 cm3 of cyclopentadiene to the KOH suspension,
stopper the tube and shake well. When a dark red brown colour develops in the tube (about
3 minutes) add the ferrous chloride solution, stopper and shake. Cool in ice if it gets
too hot. Allow the mixture to stand for 5 minutes then pour it into 200 cm3 of
cold water and filter the resulting mixture using a Buchner Funnel. After drying at the
pump for 20 minutes, transfer the solid and filter paper to a dry 200 cm3
beaker. Add 50 cm3 of 60-80% petroleum ether and heat to boiling on a
water-bath. Filter into another dry 200 cm3 beaker using a filter funnel fitted
with fluted filter paper. Concentrate the resulting orange solution on a water-bath until
crystallisation begins (reduce to 5-10 cm3). Cool in ice to complete
crystallisation.
After standing for 30 minutes, filter off the crystalline product and dry at the pump.
This last filtration may be carried out in the open laboratory. Record your yield as a
percentage based on FeCl2 and determine the M.P (this should be compared to the
literature value).
A cyclic voltammogram of ferrocene (0.1 mM)
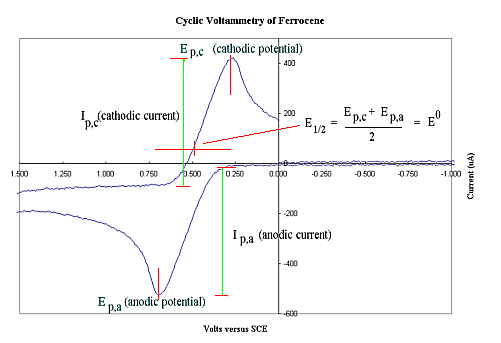
recorded in DMF using [n-Bu4N]PF6 (0.1M) as supporting electrolyte shows a reversible
oxidation wave at E1/2~0.5 Volt which indicates that at a potential slightly
higher than this (eg 0.6 V) a bulk electrolysis experiment should produce the ferrocinium
cation.
Alternatively an oxidant such as H2SO4 can be used. The ferricinium
ion [Fe(C5H5)2]+ produced is soluble in water
and may be precipitated using a large counter anion such as picrate, 12-tungstosilicate,
reineckate- or even perchlorate. In this exercise we will use 12-tungstosilicate.
Dissolve 0.5 g of ferrocene in 10 cm3 concentrated sulfuric acid; allow the
solution to stand for at least half an hour, then pour it into 150 cm3
distilled water. Stir the solution for a few minutes and filter off any precipitate. To
the filtrate add a solution of 2.5 g 12-tungstosilicic acid in 20 cm3 water
slowly with stirring. Collect the pale blue precipitate, wash with water and dry.
Ferrocene has an extensive aromatic-type reaction chemistry, which is reflected in its
name and undergoes substitutions more readily than does benzene. One example is
acetylation of both rings in the presence of a Friedal-Craft catalyst.
Alternatively, the acetylation of ferrocene can be carried out under milder conditions
using acetic anhydride in phosphoric acid to yield the mono-acetylated product.
G.A. Perkins and A.O. Cruz, J. Amer. Chem. Soc., 1927, 49, 517.
1. Propose a reaction scheme for the acetylations. The 1H nmr spectra for ferrocene and a crude mixture obtained from the mono-acetylation using acetic anhydride are provided. Record the chemical shifts (NB. TMS=0) and interpret each spectrum.
 Return to Chemistry, UWI-Mona, Home Page
Return to Chemistry, UWI-Mona, Home Page
Created Oct 1995. Last modified 27th May 1998.
URL http://wwwchem.uwimona.edu.jm:1104/lab_manuals/c21jexpt.html