Microwave Oven Reaction Enhanced (MORE) preparation of substituted stilbenediamines
The Department of Chemistry, University of the West Indies, Mona, Kingston 7, JAMAICA.
Email addresses: rjlanc@uwimona.edu.jm and pbreese@uwimona.edu.jm
Abstract
Transition metal complexes formed from stilbenediamines have been of interest for some
time. From the early work on the unusual solvent effects on the nickel complexes known as
Lifschitz's salts to more recent studies where manganese complexes have been utilised in
the chiral epoxidation of alkenes.
In an effort to shorten the preparation time of the compounds, the condensation of
benzaldehyde with ammonium acetate has been re-examined under microwave conditions.
Introduction
It was observed by Trippett in 1957 (l), that when
benzaldehyde was refluxed with ammonium acetate or ammonium propionate for three to four
hours, a highly crystalline neutral compound, N-benzoyl-N-benzylidene
meso-1,2-diamino-1,2-diphenylethane (C28H24N2O), was produced. Hydrolysis of this compound
with 70% sulfuric acid over three hours was reported to yield meso-1,2-diamino-1,2-diphenylethane
(stilbenediamine or stien).
More recently, there has been considerable interest in the use of the optical isomers of
stien (either R,R or S,S) as
precursors to catalysts used for enantiometric epoxidation of olefins (2) or as chiral Lewis acids to catalyse enantioselective
Diels-Alder reactions.(3)
The difference between these forms can be seen in Figure 1.
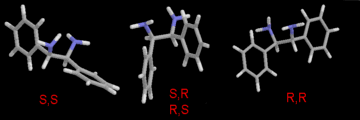 Figure
1. (enlarged GIF, 16K)
Figure
1. (enlarged GIF, 16K)
New synthetic approaches to the optically active forms have been reported recently. These
include the hydrolysis of a chiral imidazoline (4), and a new
preparation of the racemate (3) which was resolved using
existing methods (5).
As part of our studies (6) (7) on
MORE (Microwave Oven Reaction Enhanced) chemistry (8) (9) we have repeated the reaction of Trippett in a microwave oven
using benzaldehyde as well as p-chloro and m-nitrobenzaldehyde as substrates. We have been
able to produce the meso compounds in two steps, with heating times reduced to
approximately one sixtieth of those in the original paper. Products analogous to those
formed in the benzaldehyde reaction are formed rapidly from p-chloro- and
m-nitrobenzaldehyde.
Discussion
It is thought that formation of a 1,2-diphenylethylenediamine derivative under such
mild conditions may be due to a benzoin-type condensation. This view is supported by the
effect of substituents on the course of the reaction.(1).
Alternatively, the product could be formed by a condensation reaction between the aldehyde
and ammonia (generated in situ) to give the imine which could undergo radical
dimerisation. Further reaction of the resulting diradical with two molecules of
benzaldehyde would lead to the adduct as shown in Figure 2.
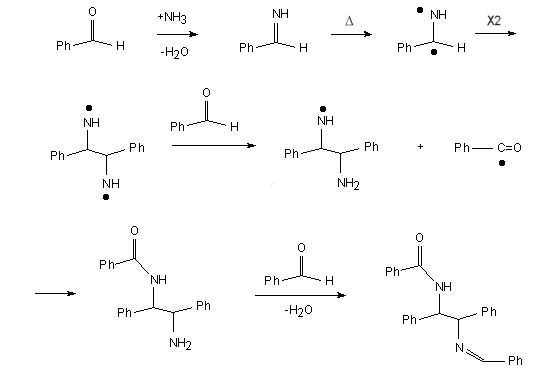
Figure 2. A possible mechanism (16K .tgf file).
The products obtained from the the microwave reactions were identical to those obtained
by classical reflux methods.
The yields in forming the adduct compared well with the reflux method, and ranged from 55%
to as high as 81% which compares to 60% as reported previously (1).
However, the hydrolysis to form the diamine generally gave yields under 50%. Using the
original conditions of 70% aqueous sulfuric acid, charring was often evident and so the
acid strength was diluted to 50%. This necessitated extending the reaction time. The lower
yields of the diamine could possibly be due to incomplete hydrolysis of the adduct.
The times used for the hydrolysis reactions were between three and four minutes, except
for the p-chlorostilbenediamine when the reactants were irradiated for five minutes.
Acknowledgements
Aspects of this work formed the basis of several undergraduate projects, for which the
authors gratefully acknowledge the assistance of: Noelle Finlay, Llewellyn Jarrett, Floyd
Beckford, Roy Porter and Maureen Wilson.
Experimental
In general the methods of preparation of the compounds were similar.
All reactions were carried out in Teflon bombs in a commercial 600 Watt microwave oven
(Sharp[TM] Carousel). The Teflon bombs were obtained from Savillex Corp. (60 cm3).
Melting points were determined on a Kofler-type micro hot stage block and are uncorrected.
Infrared spectra were recorded as potassium bromide (KBr) discs using a Perkin Elmer FTIR
1600 model spectrometer and transferred via serial cable to a PC as JCAMP-DX files.
The mass spectra were recorded at 70 eV via solid insertion in a Finnigan GC/MS quadrupole
spectrometer. Information from the 20 most prominent peaks was manually converted into JCAMP-DX format file for presentation.
Proton NMR spectra were recorded on either a Bruker AC200 or Varian T60 and were for
deuterochloroform solutions. Tetramethylsilane served as the internal standard.
N-benzoyl-N'-benzylidene meso-1:2-diphenylethylenediamine.
Benzaldehyde (15.1cm3, 15g, 140 mmol) and ammonium acetate (30.12 g, 390 mmol) were added
to a Teflon bomb which was sealed and placed in a microwave oven. The vessel was heated
for three minutes, cooled and the mixture filtered. The product was washed with ethanol,
air dried then recrystallized from toluene. M.P. 253-255 C (lit. 258-259 C). Yield 14.32g,
56%. The Mass Spectrum showed m/e at 404.
IR Spectrum (JCAMP-DX file).
The 200 MHz proton NMR (85K JCAMP-DX file) gave signals at
8.30 (s),7.79-7.24 (m), 6.71 (d) 5.55 (dd), 4.95 (d) and 1.55 (s).
meso-1:2-diphenylethylenediamine.
The adduct (5g, 12 mmol) along with 50% sulfuric acid (25 cm3) were added to a Teflon bomb
and placed in a microwave oven. The vessel was heated for 3.5 minutes and then cooled. The
mixture was extracted with ethyl acetate (3 times 30 cm3). The organic layer, which
contained unreacted starting material and other impurities was discarded. The aqueous
layer was made alkaline (pH~10) using KOH and further extracted with ethyl acetate. The
organic extract was washed with water, dried with sodium sulfate and filtered. The
filtrate was evaporated in vacuo. The product was recrystallized from methanol.
M.P. 119-120 C (lit.120.5 121.5 C). Yield 2.62g, 24%. The Mass
Spectrum showed m/e at 212.
IR Spectrum- (JCAMP-DX file)
The 200 MHz proton NMR (85K JCAMP-DX file) gave signals at
7.41 (m), 5.41 (s) and 1.47 (s).
Preparation of adduct from p-chlorobenzaldehyde.
p-Chlorobenzaldehyde (5.04 g, 36 mmol) and ammonium acetate (10.06 g, 130 mmol) were added
to a Teflon bomb which was sealed and placed in a microwave oven. The vessel was heated
for 2.5 minutes, cooled and the mixture filtered. The product was washed with ethanol and
air dried. The product was recrystallized from butan-l-ol. M.P. 251-253 C (lit. 249 C).
Yield 1.5g, 31%.
IR Spectrum- (JCAMP-DX file)
The 200 MHz proton NMR (85K JCAMP-DX file) gave signals at 8.26
(s), 7.77-7.12 (m), 6.62 (d), 5.52 (dd), 4.96 (d), 1.69 (broad-s) and 1.28 (s).
meso-1:2-di-(p-chlorophenyl)ethylenediamine.
The adduct (0.9 g, 2 mmol) and 40% sulfuric acid (4.5 cm3) were added to a Teflon bomb
which was sealed and placed in a microwave oven. The vessel was heated for 5 minutes,
cooled and the mixture extracted with ethyl acetate (3 times 30 cm3) which was discarded.
The aqueous portion was made alkaline (pH~9) using KOH and further extracted with ethyl
acetate. The organic extract was washed with water, dried with sodium sulfate and
filtered. The filtrate was evaporated using a rotary evaporator and the product
recrystallized from methanol. M.P. 140-141 C (lit 137-138 C). Yield 0.24g, 51%.
IR Spectrum- (JCAMP-DX file)
The 60 MHz proton NMR (8K GIF file) gave signals at 7.25 (Ar-H),
3.9 (N-C-H) and 1.5 (N-H).
Preparation of adduct from m-nitrobenzaldehyde.
m-Nitrobenzaldehyde (5.14 g, 34 mmol) and ammonium acetate (10.14 g, 132 mmol) were added
to a Teflon bomb which was sealed and heated in a microwave oven for 3 minutes. The vessel
was then cooled and the mixture filtered. The crystals were washed with ethanol and the
product recrystallized from butan-l-ol. M.P. 300 C (sublimes) Yield 4.01g, 81%.
meso-1:2-di-(m-nitrophenyl)ethylenediamine.
The adduct (4.01 g, 7 mmol) and 50% sulfuric acid (23 cm3) were added to a Teflon bomb
which was sealed and heated in a microwave oven for 4 minutes. The vessel was then cooled
and the organic portion extracted with ethyl acetate (3 times 30 cm3) which was then
discarded. The aqueous portion was made alkaline (pH~10) using KOH and further extracted
with ethyl acetate. The organic extract was washed with water, dried with sodium sulfate
and filtered. The filtrate was evaporated under reduced pressure and the product was
recrystallized from methanol. M.P. 184-187 C (lit. 189-190 C). Yield 0.63g, 30%.
IR Spectrum- (JCAMP-DX file)
References
[1].
S. Trippett, J. Chem. Soc., (1957), 4407-4408.
[2]. W. Zhang, J.L. Loebach, S.R. Wilson and E.N. Jacobsen, J. Am. Chem.
Soc., 112, (1990), 2801-2803.
[3]. E. J. Corey, R. Imwinkelried, S. Pikul and Y. Bin Xiang, J. Am. Chem.
Soc.,111, (1989), 5493-5495.
[4]. R. Oi and K.B. Sharpless, Tetrahedron Lett., 32, (1991), 999-1002.
[5]. O.F. Williams and J.C. Bailar, J. Amer. Chem. Soc., 81, (1959), 4464.
[6]. R.J. Lancashire and S.A. Clarke, Jamaican J. of Sci. and Tech., 4,
(1993), 32-42.
[7]. R.J. Lancashire, (1990) unpublished results. See URL
http://wwwchem.uwimona.edu.jm:1104/lab_manuals/MORE.html or here.
[8]. A.K. Bose, T. Tabei, V.S. Raju, Tetrahedron Lett.,31, (1990),
1661-1664.
[9]. D.R. Baghurst, D. Michael and D.M.P Mingos, Chem. Soc. Rev., 20,
(1991), 1-47.
Return to top of article
 Figure
1. (enlarged GIF, 16K)
Figure
1. (enlarged GIF, 16K){kind=link}